
Copyright
by
Galen Daryl Knight
1982
Dedicated to My Parents
RESOLUTION AND RECONSTITUTION OF THE NADPH-DEPENDENT
TYROSYL-PEPTIDE IODINATING ACTIVITY
FROM PORCINE THYROID TISSUE
by
Galen Daryl Knight, B.S.
DISSERTATION
Presented to the Faculty of the Graduate School of
The University of Texas at Austin
in Partial Fulfillment
of the Requirements
for the Degree of
DOCTOR OF PHILOSOPHY
THE UNIVERSITY OF TEXAS AT AUSTIN
August, 1982
ACKNOWLEDGEMENTS
The author wishes to express his gratitude to his supervising professor, Dr. D. M. Ziegler, for his guidance and support throughout the course of this study. The author also thanks Dr. L. L. Poulsen for constructive critisism and suggestions, Dr. J. R. Brown for use of his facilities in determining amino-acyl compositions, the late J. Sanford for his collaboration in purifying the hepatic monooxygenase, Dan Sulzer for preparing hepatic microsomes, Barbara Small and Dr. Mike Duffel for their respective collaboration in electrophoretic techniques and thiol-disulfide analysis, and Jane Hunter and the late Dorothy Oliver for technical assistance and encouragement.
The author also appreciates the encouragement and financial support given him by his family throughout the course of this work.
Financial support provided by Clayton Foundation Biochemical Institute is gratefully acknowledged.
Galen D. Knight
August, 1982
ABSTRACT
Thyroxine is synthesized in the thyroid from iodinated tyrosyl residues within thyroglobulin. An NADPH-dependent iodinating system has been resolved, purified, and reconstituted from hog thyroids. Optimal activity requires the following: at least two proteinaceous components, soluble factor and a microsomal monooxygenase; cysteamine or certain aminoethanethiol derivatives; nearly physiological concentrations of iodide; and a second microsomal component that has not been chemically defined. At least twelve-fold synergism is observed with the reconstituted system.
Soluble factor, the protoporphyrin IX-containing component, is probably the intact larger subunit of thyroid peroxidase. Spectral transitions in this membrane-associated protein upon exposure to tyrosyl substrates suggest an iodotransferring function for the factor. The monooxygenase resembles the hepatic microsomal FAD-containing monooxygenase (EC 1.14.13.8) electrophoretically, chromatographically, and enzymically. Since iodide and cysteamine are oxidized in reactions catalyzed by the hepatic enzyme, and other substrates for the monooxygenase inhibit NADPH-dependent iodination, this enzyme is implicated in the thyroidal metabolism of iodide. While the NADPH- dependent system retains full activity in the presence of endogenous concentrations of glutathione, peroxide-dependent iodination is completely inhibited. In addition, NADPH-dependent iodination is inhibited, but peroxide-dependent activity is stimulated by 60 µM thiocyanate. NADPH-dependent iodination is also more specific for tyrosyl-peptides than peroxide-dependent activity.
Requirements for aminoethanethiols and sensitivity of the iodinating activity to amines, guanidines, thiocyanate, and thiocarbamides suggest a mechanism involving sulfenyl iodides. Thiocyanate and perchlorate are not substrates for the hepatic monooxygenase, but they do inhibit reactions catalyzed by this enzyme. The possibility that the monooxygenase catalyzes the production of iodinating intermediates is discussed with reference to previous studies.
TABLE OF CONTENTS
I. INTRODUCTION
A. Historical Perspective
B. Formation of Iodothyronines
1. Requirements
for Iodination
C. Regulation of Iodothyronine Biosynthesis
1. Calcium
2. Sulfhydryls
3. Iodide
and the Wolff-Chaikoff Effect
D. Attempts to Purify the System
1. Loss of
Sulfhydryl-Dependency
2. Loss of
NADPH-Dependency
3. Loss of
Calcium Requirement
4. Loss of
Specificity of Iodination
5. Sensitization
to Ascorbate
6. Nonproteolytic
Solubilization of Thyroid Peroxidase
7. Chromatographic
Purification of the Peroxidase
8. Characteristics
of Purified Thyroid Peroxidase
9. Kinetic
Constants for Thyroid Peroxidase
E. Sulfenyl Iodides in Thyroidal Metabolism
1. Chemistry
of Sulfenyl Iodides
2. Iodination
F. Microsomal FAD-Containing Monooxygenase
1. Cysteamine
and Sulfenyl Iodides
2. Monooxygenase
in the Thyroid
3. Other
Activities and Characteristics of the Hepatic Monooxygenase
4. Alkylamines
and Calcium Effects on the Hepatic Monooxygenase
5. Effects
of pH and Temperature on the Monooxygenase
II. MATERIALS
A. Reagents
B. Peptides, Proteins, and Enzymes
C. Detergents
D. Peptides for Iodination
E. Preparation of Diethylaminoethyl
Cellulose Acetate
F. Preparation of Crude 4-Phosphopantetheine
G. Miscellaneous
III. METHODS
A. Assay of Glutathione and Cysteamine
B. Assays of Enzymes Influencing
Thiol to Disulfide Ratios
C. Assay of Microsomal FAD-Containing
Monooxygenase
D. Assay of Glucose Oxidase
E. Assay of Thyroid Peroxidase
F. Assay of NADPH-Dependent Iodination
G. Assay of Peroxide-Dependent Iodination
H. Assay for Iodination with Small
Peptides
I. Slab-Gel Electrophoresis
J. Protein Determination
K. Preparation of Heme Derivatives
L. Assay of Cytochrome c
Reductase
IV. RESULTS
A. Tissue Collection and Subcellular
Fractionation
1. Preparation
of Microsomes
B. Endogenous Modulators of the Iodinating
System
C. Verification of Assay for Thyroidal
NADPH-Dependent Iodination
1. Iodination
of Half-Cystinyl P-9
2. Elimination
of Peroxide-Dependent Iodination
3. Optimal
Concentrations of Glutathione and Cysteamine
4. Differentiation
with Thiocyanate
D. Resolution of Components
1. Purification
of Soluble Factor
2. Purification
of Microsomal FAD-Containing Monooxygenase
3. Elution
of the Unidentified Microsomal Factor
E. Reconstitution of Iodinating Activities
F. Properties of Purified Components
1. Soluble
Factor
2. Thyroidal
Monooxygenase
3. Unknown
Microsomal Component
G. Activities of Individual Components
1. Soluble
Factor
2. Thyroidal
Monooxygenase
H. Refinements to the Assay of NADPH-Dependent
Iodination
1. Inhibition
by Half-Cystinyl P-9
2. Iodination
of Lysylglutaryl P-9
3. Other
Aminoethanethiols
I. Oxidation of Iodide Catalyzed
by the Hepatic Monooxygenase
1. Iodide
Kinetics
2. Calcium
and Octylamine Effects
3. Cysteamine
Kinetics
4. Protection
of NADPH by Cysteamine
5. Synergism
of Cysteamine and Iodide
6. Effects
of Aminoethanethiol and Iodide on the Monooxygenase
7. Recombinant
Studies with Purified Peroxidase
V. DISCUSSION
APPENDIX
A. Humoral Modifiers of Thyroid Function
B. Uptake of Iodide by the Thyroid
1. Thyroidal
Distribution of Iodine
2. Requirements
for Uptake
3. Evidence
for Passive Diffusion
4. Evidence
for Active Transport
5. Concentration
of Analogous Anions
C. Factors Influencing Iodide's Accumulation
1. Catecholamines
2. Acetylcholine
3. Thyrotropin
4. Other
Endocrine Modulators
5. Xenobiotic
Compounds
VI. REFERENCES
LIST OF TABLES
LIST OF FIGURES
ABBREVIATIONS
The following abbreviations are used in this dissertation:(-) - inhibitor
(+) - activator
AMP - adenosine monophosphate
cAMP - adenosine-3',5'-cyclic-monophosphate
CPM - counts per minute per assay
DMA - N,N-dimethylaniline
DNFB - 2,4-dinitro-1-fluoro-benzene
EDTA - ethylene-diamine-tetraacetic acid
FAD - flavin adenine dinucleotide
FMN - flavin adenine mononucleotide
GSH - glutathione
GSSG - glutathione disulfide
HMP Shunt - hexose-monophosphate shunt
Ki - Michaelis inhibition constant
Km - Michaelis dissociation constant
MMI - N-methyl-2-mercapto-imidazole (methimazole)
NAD+ and NADH - oxidized and reduced nicotinamide adenine dinucleotide
NADP+ & NADPH - oxidized and reduced nicotinamide adenine dinucleotide
PTU - 6-propyl-2-thiouracil
[S]½ - concentration of substance necessary to half-saturate an effect
T3 - 3,5,3'-triiodothyronine
TPO - thyroid peroxidase
TSH - thyrotropin
Z - benzoyl-
HISTORICAL PERSPECTIVE
Fascinations with localized swellings of the neck, no doubt, antedate recorded history; references to goitre, the most obvious of thyroidal disorders, are construed from some of our oldest decipherable records. Herbal remedies for the malady, such as the seaweed Sargassum, are prescribed in Chinese literature dating back to 2700 B.C. (Shen-Nung) [1]. Since that time, burnt sponge from 1600 B.C. [2], surgery or salt from 1500 B.C., and animal thyroid from 500 A.D. (Shen Shifan), have all been used to reduce swellings of the lower neck. These treatments, with the exception of burnt sponge, were sporadically practiced into the twentieth century [1].
Iodine was first detected in seaweed in 1811 (Courtois), sea sponges in 1819 (Fyfe), and in the thyroid itself in 1895 (Baumann). Proust and Coindet, however, as early as 1816 and 1820, respectively, were recommending iodine for treatment of goitre [1]. The active form of iodine in the thyroid, thyroxine, was obtained in pure form in 1914 by Kendall [3]. Chemical synthesis of the hormone was accomplished twelve years later by Harington. During this same period, largely through the efforts of Marine and Kimball, iodine became acceptable in goitre prophylaxis [1].
Iodine also proved useful in the treatment of hyperthyroidism. Plummer, as early as 1923, prescribed iodine to moderate the hyperactive thyroid [2], thereby reducing the chance of thyroid storm during later surgery [4]. The beneficial effect of high concentrations of iodine in diminishing, rather than augmenting, the hormonogenic efficiency of the thyroid was later studied by Morton, Chaikoff, and Rosenfeld [5]. This inhibition by iodide, coined the Wolff-Chaikoff Effect [6], has been used to compare various reconstructed systems with the biosynthetic machinery in the intact thyroid [7].
Xenobiotic therapy for hyperthyroidism evolved
from epidemiological studies. Characterization of various dietary and environmental
goitrogens led to the observation that members of the thiocarbamide class
of chemicals effectively perturb the synthesis of thyroxine and triiodothyronine
[1,2,3,7].
Included in this group are derivatives of thioacetamide, thiourea, mercaptoimidazole
(imidazolethione), and thiouracil. Several of these compounds proved useful
in probing the biosynthetic pathway for thyroxine: thiocyanate, propylthiouracil,
N-methyl-2-mercaptoimidazole, and thiourea [7]. Toxicities
of the drugs and their various metabolites have limited the therapeutic
value of these agents. Consequently, research on the thyroid has diverged
in two directions: studies of the hormonal regulation of the thyroid gland,
and studies of the intrathyroidal production of thyroxine.
FORMATION OF IODOTHYRONINES
The synthesis of thyroid hormones is an oxidative process. Cell free preparations of thyroid catalyze both the incorporation of iodide into tyrosyl residues of proteins, and later coupling of iodinated residues to form proteinaceous triiodothyronine and thyroxine, as the following proposed sequence of reactions illustrates [7]:
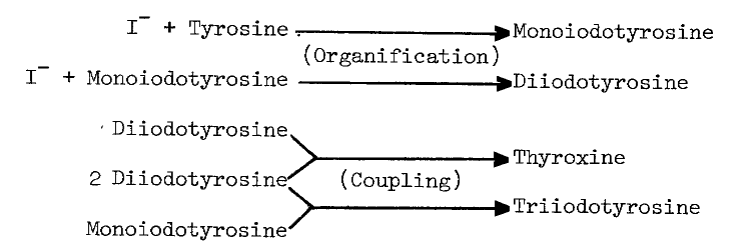
These activities sediment predominantly in
the microsomal fraction at about 10,000 to 60,000xg, but a heavier fraction,
mitochondria, does contain appreciable activity [8,9].
Attempts to purify components of this pathway further have met with limited
success, for even the most homogeneous preparations of thyroid peroxidase
are seldom more than half pure. One
preparation removed from microsomal membranes by proteolysis is estimated
to be 80 percent pure, but contains at least two other proteins [10].
A comprehensive review of thyroid peroxidase and its activities has recently
been compiled [7].
Requirements for Iodination. Oxygen is essential for the organification of iodide. In homogenates, the requirement differs from that for the accumulation of iodide by slices; accumulation and organification of iodide are half-saturated by 5 and 20 percent oxygen, respectively [11,12].
Reduced pyridine nucleotides, NADPH in particular, stimulate iodination in microsomes [7,13]. At the same concentrations, 0.1 mM NADH sustains 80 percent of the iodinating activity observed with NADPH-stimulated microsomes [13]. As little as 10 µM ascorbate completely inhibits oxidation of NADH catalyzed by the particulate fraction [14], but allows most of the iodination [14,15,16]. Purification of the particulate fraction may alter interaction of reduced pyridine nucleotides with the iodinating system, for NADH is slightly inhibitory in homogenates [12].
Differential effects in homogenates and microsomes are also observed in response to oxidized pyridine nucleotides. NADP+ suppresses iodination in homogenates [12], but stimulates iodination in microsomes [13] and dialyzed homogenates. NAD+ enhances iodination in both preparations, but inhibitory concentrations of NADP+ are prerequisiste for maximal NAD+ effectat least in homogenates [12]. In microsomes NAD+ is more effective than NADP+ in enhancing iodinating activity [13].
Glutathione is involved in the response of iodination to pyridine nucleotides. Glutathione (GSH) inhibits iodination more than the disulfide (GSSG), but NADP+ and GSSG in combination are nearly as inhibitory as GSH alone. Removing glutathione from the system, either by dialysis or sedimentation of microsomes, reverses inhibition by NADP+, an effect restored by GSSG. The action of GSH is attributed to reductive removal of peroxides and intermediary iodines essential to iodination, as illustrated by the following scheme [12]:
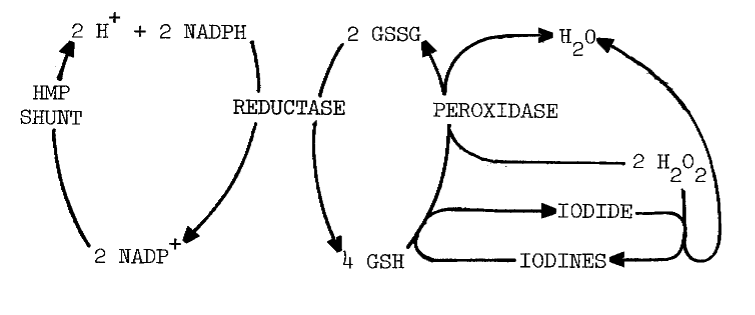
Alternately, the disulfide may maintain NADP+ in the oxidized formone that inhibits, rather than stimulates, iodination. Adding NADP+ to homogenates does inhibit iodination [12] until the 0.7 mM NADP+ and possibly GSSG are reduced. No such inhibition is observed when NADPH is added to microsomes [13]. Responses of these two preparations cannot be directly compared, since microsomes lack glutathione present in homogenates.
Flavin nucleotides, FAD and FMN, stimulate
iodination in both homogenates and microsomes. The effect is dependent
upon the presence of oxygen, and NADPH or light [12,13,17].
Dialysis has little effect upon the flavin requirement, so flavin seems
to be tightly associated with some component of the system [13].
The microsomal component is also heat labile [17]. Taken
together, these results indicate that flavin serves as coenzyme for a specific
microsomal enzyme catalyzing some step in the iodinating process.
Catalase prevents the flavin-dependent stimulation and inhibits up to 75 percent of the original activity [13]. The effect of catalase is diminished by FMN, especially at higher pH (8.0). FMN also diminishes the requirement for oxygen. A similar decrease in the requirement for oxygen occurs as pH is increased [17]. Presumably, flavin reduced either by light or NADPH reacts with oxygen to form hydrogen peroxide and oxidized flavin, as the following scheme indicates [12]:
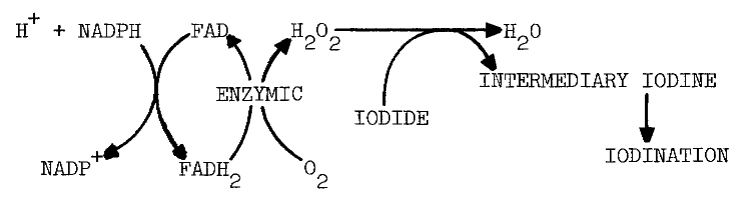
The peroxide then oxidizes iodide and eventually produces iodothyronines in reactions catalyzed by thyroid peroxidase. Peroxide and peroxide-generating systems do circumvent the requirement for NADPH in cell-free systems [7]. Problems with such substitutions will be examined later.
Calcium also profoundly influences NADPH-dependent iodination. Concentrations approximating those of plasma (1 mM) are optimal. Doubling or halving the concentration of calcium ions causes precipitous losses in particulate iodinating activity [13].
Thyroglobulin and its precursors are primary targets for iodination. In untreated animals, iodination and coupling of newly synthesized thyroglobulin precede that of thyroglobulin stored in the lumen [18]. Newly synthesized thyroglobulin is associated with the particulate fraction [19,20] and is iodinated soon after synthesis. This explains iodinations observed in isolated cells practically devoid of colloid [21]. The colloid, therefore, serves as a reservoir of partially iodinated thyroglobulin and iodide for use when demand on hormonogenesis is high.
As thyroglobulin is iodinated it becomes progressively more dense [22]. Bound iodide [7,22] and conformational changes apparently contribute to this maturation process. A more compact protein probably arises through changes in tyrosyl and cysteinyl residues. The pKa of tyrosine (10.13) drops as it is iodinated first to monoiodotyrosine (8.20) and later to diiodotyrosine (6.36). Since ionization at physiological pH is appreciable only when tyrosyl residues are fully iodinated, such a change may initiate the maturation process. The more thoroughly iodinated thyroglobulin is also enriched in disulfides [23] that provide further conformational constraints. Folding of thyroglobulin into a faster sedimenting conformation is thought to facilitate coupling by bringing iodotyrosyl residues into proper juxtaposition [7].
Both iodination and coupling favor amino-acyl
sequences containing adjacent glutamyl and tyrosyl residues [24,25].
The sequence (-alanyl-seryl-tyrosyl-glutamyl-aspartate-) is the most hormonogenic
of any isolated from thyroglobulin [24]. In contrast
to coupling, iodination occurs more readily on denatured, or unfolded,
thyroglobulin [13]. Other proteins and peptides are iodinated
[7,23]
probably by virtue of similar sequences positioned such that iodination
is favored and coupling is excluded.
REGULATION OF IODOTHYRONINE BIOSYNTHESIS
Calcium. Thyrotropin is the single most important modulator of
thyroid function. However, several of its effects are mimicked by neurotransmitters,
acetylcholine and catecholamines (Table 9 in the Appendix).
Of the responses to these stimuli, increases in cytosolic calcium correlate
most closely with iodinating activities. High concentrations of calcium
in the medium, or low concentrations in the presence of calcium ionophores,
mimic the effects of acetylcholine [26]. Removing calcium
from the medium prevents TSH-dependent stimulation of two activities: iodination
of protein, and production of NADPH by the hexose-monophosphate (HMP) shunt
[27,28].
These effects of calcium are independent of the cyclic AMP response to
thyrotropin [26,28]. Ca++-dependency
is, therefore, a distinguishing characteristic of systems metabolizing
iodide. Indeed, accumulation and organification of iodide are optimal at
0.9 mM [29] and 1 mM [13] calcium,
respectively. Circulating levels of calcium are higher at 1.16mM [4,30].
Since Ca++-dependency is demonstrable with
intact cells [29] and microsomes
[13],
only two aspects of this iodinating system remain unresolved: the stimuli-induced
translocation of calcium in the cell and the microsomally catalyzed iodination
of thyroglobulin.
Sulfhydryls. Certain compounds stimulate the uptake of iodide by the thyroid, in vivo: cysteamine, cystamine, and penicillamine [31]. Stimulation occurs only when organification of iodide has been previously blocked by thiocarbamides. In untreated thyroids cysteamine mimics, albeit weakly, the action of thiocarbamides. Concentrations of methimazole or cysteamine required to half-inhibit organification are 1.4 and 71 µM, respectively. Unlike the thiocarbamides, however, neither cysteamine nor cystamine is goitrogenic. Cysteamine is concentrated more efficiently by the thyroid than by any other organup to eight fold [32]but thyroidal accumulation of propylthiouracil and methimazole is nearly 500 and 1,000 fold, respectively [33,34].
The precise mechanism through which aminoethanethiols exert this stimulation is unknown. Cysteamine and cystamine do stimulate a ouabain-sensitive ATPase activity in the thyroid, but no close correlation exists between this activity and the accumulation of iodide [28]. Alternately, cysteamine and iodide may be oxidized to a sulfenyl iodide, thus establishing a metabolic gradient into the thyroid. The feasibility of such an oxidation in the presence of thiocarbamides has been questioned as has the stability of the resulting sulfenyl iodide [32]. A related aminoethanethiol, the following modified penicillamide, does form a sulfenyl iodide of atypical stability:
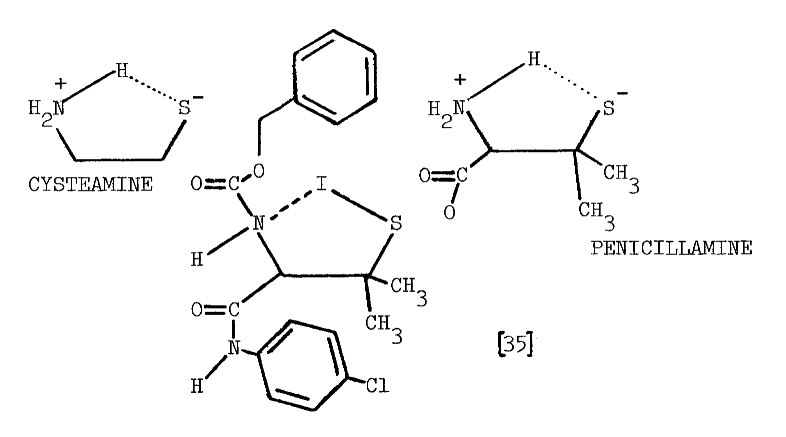
Enzymes catalyzing the formation of these labile compounds are not known, so specificities for various sulfhydryl substrates have not been established.
In contrast to the reported deleterious effects of glutathione on the iodination of protein, low concentrations of certain thiols stimulate incorporation of iodide into protein; 0.1 mM cysteine or penicillamine stimulate the particulate-catalyzed iodination of protein by 61 and 78 percent, respectively. The following structural considerations indicate that these compounds are derivatized aminoethanethiols:
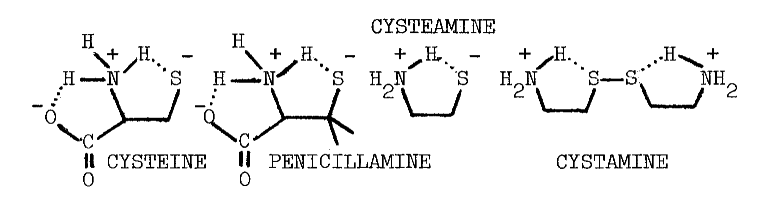
Higher concentrations of these compounds inhibit iodination [36] in a manner not unlike cysteamine's inhibition of organification [32]. The concentration of cysteine half-inhibiting iodination is four times that required to obtain the same effect using penicillamine, and nearly twenty times the 71 µM observed using cysteamine [32,36]. These rather specific responses to aminoethanethiols, and the virtually identical requirements for calcium in the two systems, provide the most convincing evidence that accumulation and organification of iodide are driven by a common mechanism.
Iodide and the Wolff-Chaikoff Effect. Iodide has long been known to suppress its own accumulation and incorporation into iodothyronines [5,6,7]. More than 90 percent of iodide accumulation and organification is inhibited in thyroids of rats exposed to 0.1 mM iodide. This is true in vivo if rats are maintained on a low-iodine diet, and in vitro [37]. Inhibition is probably not limited to exogenous iodide, since enough iodide can arise from thyroglobulin to interfere with accumulation [38]especially after acute exposure to TSH [31,39,40,41,42,43]. Intrathyroidal concentrations provide a better index of iodide's effect on its own metabolism. In slices or isolated cells from bovine thyroids, organification into iodotyrosines and iodothyronines is optimal at 60 µM intrathyroidal iodide, half-inhibited at 0.3 mM, and completely inhibited at 0.9 mM iodide [44].
Several mechanisms through which iodide influences its own metabolism have been explored. Inhibition is secondary to the formation of organic iodine(s) in most cases, and is associated with an increase in the [S]½ for iodide in the accumulating system. Thus, by limiting uptake, the thyroid adapts to the higher exogenous concentrations of iodide [45]. The organic iodine is not triiodothyronine or thyroxine [26,45,46]. The iodine inhibiting uptake may be an intermediate in the iodination of protein, since higher concentrations of prethyroglobulin make demonstration of the Wolff-Chaikoff Effect more difficult [7].
Iodide does depress the iodinating activity of thyroid peroxidase, but only in highly purified preparations and only at concentrations of iodide, 5.2 mM, in excess of those having a similar effect in intact tissue, 0.3 mM [7,44,47]. Iodide, apparently, reduces a peroxidase-oxidized iodide complex to form free peroxidase and iodine, as the following scheme illustrates [48]:
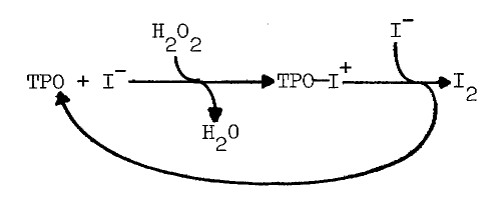
The complex has been isolated and shown to iodinate the tripeptide, glutamyl-tyrosyl-glutamate [49]. Generation of iodine is associated with less efficient iodination, even though iodine is a potent iodinating agent [48].
At lower concentrations of iodide or thiocyanate the coupling of N-acetyltyrosinamide is stimulated [50]. Iodide or thiocyanate is also required for peroxidase-catalyzed oxidation of thiourea, propylthiouracil, and methimazole [51]. Peroxidase-catalyzed coupling of N-acetyltyrosinamide is optimal at 0.5 mM concentrations of either anion. Apparent Km for stimulation of coupling are 0.058 or 0.1 mM for iodide and thiocyanate, respectively [50]. Although this approximates the Wolff-Chaikoff Effect in intact tissues, the relationship does not always hold; when thyroglobulin is the substrate, iodide stimulates peroxidase-catalyzed iodination at the expense of coupling. Iodination of thyroglobulin is half-saturated by 60 µM iodide [7,52].
Since no Wolff-Chaikoff Effect is demonstrable in crude peroxide-dependent systems, iodide is thought to inhibit its own metabolism in vivo by suppressing generation of oxidative equivalents. One way in which this may be accomplished is by depletion of reduced pyridine nucleotides [7]. This reportedly occurs without prior oxidation of iodide, since propylthiouracil and N-methylmercaptoimidazole do not affect depletion of reduced pyridine nucleotides [47,53]. Although loss of NAD+ and NADH exceeds that of NADP+ and NADPH, the distribution of these nucleotides in the thyroid indicates that NADPH and NADP+ are preferentially depleted [53]. This depletion is consistent with the greater interaction of NADPH with the iodinating system [7,12,13]. NADPH-cytochrome c reductase is touted as the most likely NADPH-dependent source of peroxide in thyroidal microsomes. Iodide, however, does not inhibit iodination in thyroidal particulate fractions [7], even when the system is supplemented with NADPH, NADPH-cytochrome c reductase, and peroxidase [54]. Iodide's inhibition of its own metabolism is, nonetheless, demonstrable in an analogous system. In the presence of copper, iodination of tyrosine catalyzed by homogenates of the submaxillary gland is half-saturated at 13 µM, optimal at 42 µM, and half-inhibited at 0.56 mM iodide [55]. Factors other than the NADPH-cytochrome c reductase system are apparently required.
A somewhat more satisfactory explanation for
the Wolff-Chaikoff Effect evolves from studies of thyrotropin's action
on the thyroid. More dramatic responses to thyrotropin are observed in
animals maintained on a low-iodine diet [56]. Iodide
inhibits many of these responses by suppressing increases in cyclic AMP
[43,45,47,56,57].
Since iodide does not alter thyroidal activities of phosphodiesterases,
the effect is attributable to suppression of synthesis [47,56,57].
Dibutyryl-cyclic AMP does circumvent iodide's inhibition of cyclic AMP-dependent
responses [47]. Inhibition may be observed only upon
metabolism to a specific organic iodine, since methimazole prevents inhibition
of adenylate cyclase activity [43,47,57].
As the system is purified, iodide inhibits more effectively, indicating
an inhibitor that is either lipid-soluble [57] or labile
to some cytosolic component. Other evidence indicates the organic inhibitor
is neither soluble nor freely diffusible [46]. The ability
to form this organic inhibitor is lost in long term culture of thyroidal
cells [46,57] or upon exposure to thyroid
hormones [31]. Cultured cells also lose their ability
to accumulate and organify iodide [46]. Peroxide or peroxide-generating
systems restore iodide-sensitivity to cyclic AMP-dependent responses [46,57].
As before, formation of this organic iodine is dependent upon an unidentified
generator of oxidative equivalents.
ATTEMPTS TO PURIFY THE SYSTEM
Although there is evidence that other components profoundly influence iodination, most work on this system revolves around thyroid peroxidase [7]. The iodinating activity isolated as peroxidase bears little resemblance to that in intact tissue and microsomes. In addition to the Wolff-Chaikoff Effect discussed in the previous section, many other responses of the iodinating system cannot be duplicated with partially purified peroxidase.
Loss of Sulfhydryl-Dependency. Stimulation of iodinating activity by aminoethanethiols occurs only in intact tissue or in the particulate fraction [31,32,36]. Once solubilized from the membrane iodinating activity is inhibited by cysteine [14,36] and penicillamine [36]. Loss of penicillamine-dependent iodination occurs even in intact particulate fractions [9]. Glutathione usually inhibits iodinations catalyzed by thyroidal preparations [9,12,14,16,58]. One third of the iodinating activity in the particulate fraction is insensitive to glutathione and influenced even less by penicillamine. Mutual exclusion of sulfhydryls and endogenously generated peroxide may be partly responsible for this inhibition. Cysteine under these conditions proves inhibitory [9]. Losses of other components-those essential to sulfhydryl-dependent iodination-are also apparent, for the same concentration of penicillamine in other particulate preparations nearly doubles iodinating activity [36].
Sulfhydryls may also inhibit iodination by reducing essential disulfides within the peroxidase, thereby irreversibly destroying activity [7]. This may be especially true for the partially purified peroxidase, for disulfides may stabilize peroxidase cleaved during proteolytic solubilization. Alternately, microsomal proteins essential to peroxidase activity may associate through disulfide bridges [10].
Loss of NADPH-Dependency. Solubilization of the peroxidase from particulate membranes changes the stimulatory action of reduced pyridine nucleotides into an inhibitory one [7,14,36,59]. NADPH- and NADH-dependent iodinating activities remaining in proteolytically solubilized preparations are thermally labile, even at 4°C [59]. Solubilization, then, changes an iodinating activity dependent upon reduced pyridine nucleotides to one that utilizes only peroxide. Peroxide-dependent iodination is more thermally labile than other peroxidase activities [60], but less labile than iodinating activity in unsupplemented preparations [11,15,59,61]. A portion of the particulate iodinating activity most sensitive to heat (1 minute at 650 C) is quantitatively indistinguishable from that insensitive to glutathione [9]. Unsupplemented activity is also nearly one order of magnitude more sensitive to sonication than peroxide-dependent iodination [15].
Loss of Calcium Requirement. Along with the loss in NADPH-dependency,
solubilized preparations become insensitive to calcium [36,62].
Iodination catalyzed by particulate fractions is stimulated by calcium
only when NADPH is present [13].
Loss of Specificity of Iodination. Prior to proteolytic solubilization, thyroidal preparations catalyze the iodination of a particulate-associated protein thought to be a precursor of thyroglobulin [13,15,18,19,20]. Upon solubilization with detergents and proteases, the system loses its specificity and iodinates albumin, casein, fibrinogen, lysosome, peptides, and even tyrosine, monoiodotyrosine, and lipids [7,15,16,48,49,54,59]. Since adding peroxide to intact particles mimics the effect [15], loss of specificity in the iodinating reaction is attributable to the deleterious effect of peroxide. Solubilization with detergents alone should extract the intact generating system and peroxidase, and circumvent the need for peroxide, but NADPH-dependent iodination becomes thermally labile upon exposure to deoxycholate [9,59].
Sensitization to Ascorbate. Ascorbate does not influence the uptake of iodide by the thyroid [31]. As the particulate fraction is purified, though, ascorbate becomes more inhibitory of iodinating reactions [14,15,16].
Nonproteolytic Solubilization of Thyroid Peroxidase. The peroxidase
is released from the membrane at high pH (10.5) and by organic solvents
or detergents [7, 63]. Solubilization
by nonionic detergents is also pH-dependent; at pH 6.4 no peroxidase is
solubilized, while solubilization is virtually complete at pH 8.4 [63].
Chromatographic Purification of the Peroxidase. Chromatography on diethylaminoethyl cellulose is widely used to purify the peroxidase. At pH 7.0 to 7.2 and low ionic strength, the peroxidase adsorbs to diethylaminoethyl cellulose. Linear gradients of KCl (0 to 50 mM) elute a proteinaceous fraction absorbing at 410 nm [16,64]. Higher concentrations of salt elute material absorbing at 410 nm, but lacking peroxidase activity [16].
Characteristics of Purified Thyroid Peroxidase. 44 kg of porcine thyroids yields 43 mg of purified thyroid peroxidase. Although the peroxidase is thought to be at least 80 percent pure, it contains only 0.65 moles of heme per mole of holoenzyme. Disulfides stabilize two subunits of molecular weights, 24,000 and 60,000, in a holoenzyme of 87,000 [10].
Crude preparations absorb maximally at 409 nm when oxidized and at 423 nm when reduced [7]. A shoulder at 430 nm is also seen in the reduced spectra. Carbon monoxide blue-shifts the reduced spectra with an increase in absorbance at 420 nm [65]. Weak absorbance from 522 to 527 and from 560 to 562 nm corresponding to reduced cytochrome is also observed [7].
More purified preparations absorb maximally at 413 nm. The millimolar extinction coefficient at this wavelength is approximately 114. Slight absorbance at 500, 543, 586, and 686 nm is also observed. Ratio of absorbance at 410 nm to 280 nm, in the absence of other hemoproteins, gives an approximation of purity; values ranging from 0.42 [66] to 0.55 have been reported. The pyridine-hemochromogen of the purified peroxidase displays maximal absorbance at 420, 562, and 590 nm, and a shoulder at 401 nm. Reduction increases absorbance of this derivative at 422, 527, and 562 nm and diminishes absorbance at 455 and 590 nm. Comparison of this spectra with the reduced pyridine-hemochromogen of horse radish peroxidase suggests that the chromophore is not ferriprotoporphyrin TX, since maxima in the latter spectra occur at 420, 525, and 557 nm [10]. Chromophore extracted into methylethylketone, nonetheless, displays a spectrum identical to protoporphyrin IX [7].
The peroxidase contains approximately 10 percent carbohydrate [10], a property exploited in the purification of intact peroxidase. Concanavalin A covalently coupled to agarose is used to purify a detergent-solubilized peroxidase. The absorbance of a 1 mg per ml protein solution at 410 nm, 0.078, is the highest reported for any nonproteolytically solubilized peroxidase. [65].
Kinetic Constants for Thyroid Peroxidase. Kinetic constants for the peroxidase may be a contradiction in terms. Many of the discrepancies are attributable to differences in preparations of the peroxidase. Choice and concentrations of supporting substrates also influence constants. These values have been recently reviewed [7]. [S]½ values for iodide with the native substrate, thyroglobulin, are fairly consistent at 72 to 100 µM [10,64], while those with tyrosine fluctuate twenty fold [7]. [S]½ values for tyrosyl substrates may increase as tyrosyl residues are iodinated, since the apparent Km's for tyrosine and monoiodotyrosine are 82 and 100 µM, respectively [49,54]. [S]½ for glutamyl-tyrosyl-glutamate, a tripeptide that effectively prevents formation of periodide, is 3 µM [49].
Concentrations of peroxide half-saturating
the system vary even more widely than those for iodide with tyrosine as
acceptor [7]. The diversity of these constants indicates
that changes in the thyroidal iodinating system may occur during purification.
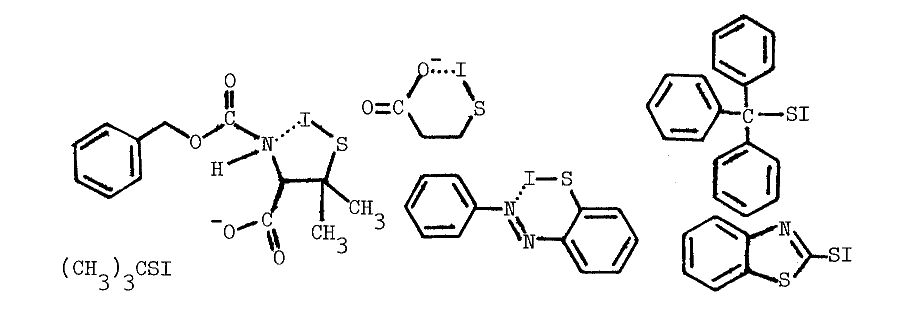
SULFENYL IODIDES IN THYROIDAL METABOLISM
Chemistry of Sulfenyl Iodides. Sulfenyl iodides form sulfenamides with amines, mixed disulfides with sulfhydryls, nitrogen with azide, and iodine and the disulfide when irradiated, as indicated in the following reactions [67,68,69,70]:
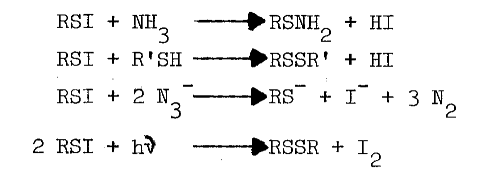
Thiols capable of forming thione-tautomers react more rapidly with sulfenyl iodides; thiouracil, thiourea, methimazole, mercaptoethanol, and glutathione are, in the order listed, less and less reactive [69]. Polar solvents destroy sulfenyl iodides by dissociating them, as follows, into the sulfenium ion and iodide:
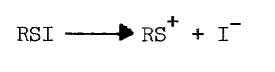
Nonpolar environments and intramolecular hydrophobic and nucleophilic groups apparently stabilize the following representative sulfenyl iodides [67,68]:
Iodination. For various reasons sulfenyl iodides are proposed as intermediates in the iodination of tyrosine. Only proteinaceous sulfenyl iodides are considered to be stable enough to be of physiological significance [7,32,71]. The iodinating system, however, is microsomal. Since iodination occurs most readily on particulate-associated precursors of thyroglobulin, and in the presence of penicillamine or cysteine [8,9,13,17,18,19,20,37], the non-polar environment of the membrane may sufficiently stabilize lipophilic sulfenyl iodides. One small sulfenyl iodide, N-carbobenzoxy-(p-chlorophenylamido)-S-iodopenicillamide, is stable for several days in dichloromethaneeven when stirred vigorously with water. The compound is sensitive to UV irradiation, for spectral loss at 430 nm is associated with a color change from orange to violet and decomposition of the sulfenyl iodide to iodine and disulfide [68]. Lipophilic sulfenyl iodides, especially those derived from aminoethanethiols, are likely iodinating intermediates at particulate interfaces.
Environments favoring the formation and longevity of sulfenyl iodides are seldom employed in studies of thyroidal iodinations. The proper reducing environment ensures a continuous supply of sulfhydryl precursors without reductively jeopardizing the sulfenyl iodide. Glutathione and ascorbate, 0.3 and 1.2 mM respectively, create such an environment in the thyroid [14]. These reductants are routinely excluded from studies on iodinating systems due to their well-documented, deleterious effect on peroxide-dependent iodination [12,14,15,16,58,65]. Thyroidal glutathione reductase normally catalyzes the reduction of glutathione disulfide by NADPH, but this system is also removed with the cytosol in preparing particulate fractions [72,73].
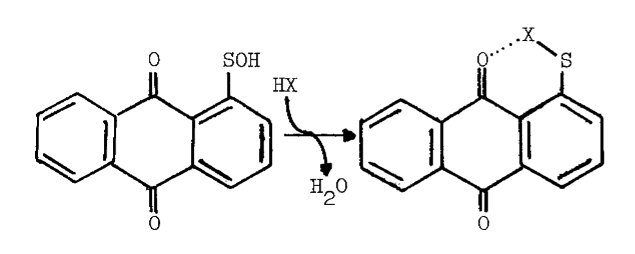
Peroxide at physiological pH oxidizes any residual sulfhydryls in reactions catalyzed by metals and glutathione peroxidase [73]. Curiously, an unidentified organic iodine produced in the particulate system disappears from the lipid fraction when peroxide is added. A concommitant loss in the specificity of iodination is observed; normally only particulate proteins and a few phospholipids are iodinated, but in the presence of peroxide, other lipids and serum proteins become good substrates for iodination [15]. The deleterious effect of peroxide is also observed in more purified preparations. High concentrations of peroxide decrease the lag period between peroxide-dependent iodination and peroxide-dependent coupling, thereby generating less thyroxine. The peroxide-induced transition from iodination to coupling is associated with completely iodinated thyroglobulin and a rapid rise in the absorbance of the system at 430 nm. Preincubation with peroxide doubles the amount of iodide needed to half-saturate peroxide-dependent iodination [52], and produces an increase in the absorbance of the partially purified peroxidase at 430 nm. Endogenous reductants may be involved in the formation of this intermediate, since spontaneous regeneration of the original spectrum is accelerated by iodide (5 µM) or ascorbate (100 µM) [65,74]. Although changes in the prosthetic group of the peroxidase are invoked to explain these phenomena [52,65,74], the observations are not inconsistent with the synthesis and degradation of sulfenyl iodides.
Methimazole (10 µM) also prevents formation of intermediates absorbing at 430 nm in reactions catalyzed by thyroid peroxidase [66]. Thione-forming thiocarbamides are contraindicated in studies potentially involving sulfenyl iodides due to the following reactions [69]:
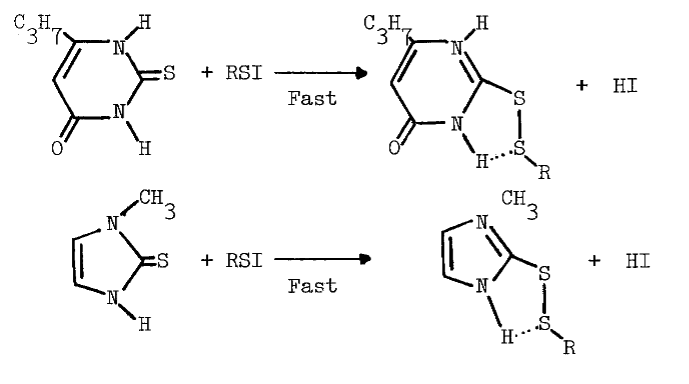
The reaction is observed with hydrogen chloride and bromide [77], and probably occurs with hydrogen iodide. The sulfenic acid of cysteamine may be held in a conformation favoring sulfenyl iodide formation, but resisting further electrophilic oxidation to the sulfenic acid:
MICROSOMAL FAD-CONTAINING MONOOXYGENASE
Cysteamine and Sulfenyl Iodides. Sulfenic acids are the initial products of thiol oxidations catalyzed by the hepatic FAD-containing monooxygenase. Since cysteamine is the only known endogenous substrate for the hepatic enzyme [78], the sulfenic acid of this aminoethanethiol is a likely physiological source of sulfenyl iodides.
Unlike intermediates in the oxidation of other thiol substrates, the sulfenic and sulfinic acids of cysteamine cannot be detected with trapping agents in reactions catalyzed by the hepatic monooxygenase. The disulfide, cystamine, is the only demonstrable product. In reactions catalyzed by thiol-disulfide isomerases, cystamine then oxidizes proteinaceous sulfhydryls to disulfides [79,80]. Presumably, the sulfenic acid remains on the enzyme until the disulfide is formed. Double reciprocal plots of rate versus concentration of cysteamine are linear at high concentrations, indicating that interaction of only one mole of cysteamine is rate-limiting [79]. In accordance with the release of the dicationic disulfide, other diamines do not interact with the monooxygenase [81].
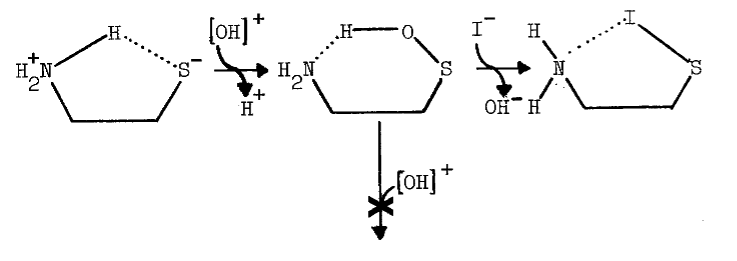
The tenacity with which the monooxygenase interacts with cysteamine is not surprising, since the aminoethanethiol is well-suited chemically for interaction with microsomal components. Protonation of the amino group effectively increases the acidity of the thiol group [82]. An appreciable amount of cysteamine, therefore, exists as the zwitterion at physiological pH, as the following scheme indicates [82,83]:
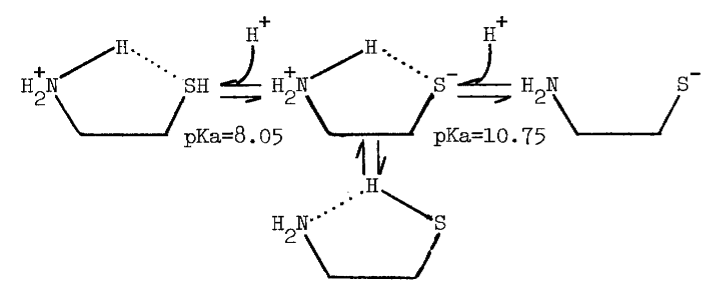
The zwitterion is in equilibrium with an uncharged species of considerable solubility in organic solvents.
Monooxygenase in the Thyroid. Monooxygenase activity and antigenicity are detectable in thyroidal microsomes. Reported N-oxygenation of N,N-dimethylaniline (0.021 nmole/minute/mg protein [84]) is much slower than expected, based upon the amount of antigen actually present [85]. Theoretically, activity in thyroidal microsomes should exceed 0.6 nmole/minute/mg protein. Although these values are lower than for microsomes from other tissues [84], antigenicity is localized in the apical membrane [86] immediately adjacent to thyroxine-containing colloid [87].
The monooxygenase may be important to thyroidal metabolism, since nearly all antithyroidal agents tested are substrates for the hepatic monooxygenase. In the absence of reduced glutathione (GSH), which reduces sulfenates, many of the antithyroidal agents are oxidized a second time to the sulfinate [88]. Nonenzymic hydrolysis of the sulfinate of methimazole yields sulfite and N-methylimidazole in the following reaction [89]:
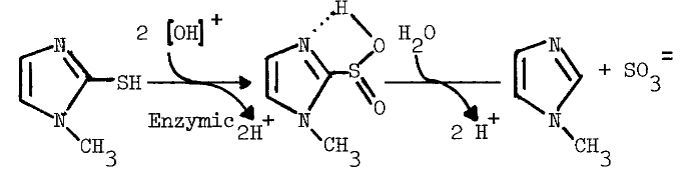
The monooxygenase catalyzes nearly all of the metabolism of methimazole in the liver [89]. Desulfuration of methimazole and other antithyroidal agents in the thyroid provides additional evidence that the monooxygenase is present in this tissue [90,91]. Although [S]½ values for the hepatic enzyme indicate that antithyroidal agents are good competitive inhibitors of monooxygenase-catalyzed reactions [89], the role of the monooxygenase in the synthesis of thyroid hormones has not been explored.
Other Activities and Characteristics of the Hepatic Monooxygenase. Iodide is also substrate for the purified hepatic monooxygenase. The product of this reaction, like iodine, irreversibly oxidizes NADPH. Monitoring the iodide-dependent disappearance of NADPH, an [S]½ value for iodide is obtained: 14.5 mM in the presence of 5 mM MgCl2 [92]. Purified hepatic monooxygenase catalyzes the N-oxygenation of a variety of tert- and sec-alkyl or arylamines, some primary arylamines, 1,1-disubstituted hydrazines [93], and disubstituted N-hydroxylamines. N,N-dimethylaniline (DMA) is a particularly good substrate with a [S]½ of less than 3 µM [94].
Thiocarbamides, thioamides, thiols, and sulfides are also substrates [79,88,89]. [S]½ for N-methyl-2-mercaptoimidazole (MMI) is 5.2 µM. The monooxygenase (EC 1.14.13.8, dimethylaniline monooxygenase) clearly is a mixed-function oxygenase, and to avoid substrate-related confusion in nomenclature is now referred to as microsomal FAD-containing monooxygenase [78].
As its name implies, the hepatic enzyme contains
flavin-adenine dinucleotide (FAD). Oxygen and reduced pyridine nucleotides,
especially NADPH, are essential substrates. With methimazole or trimethylamine
the mechanism of monooxygenase-catalyzed reactions is now known to be ordered.
Michaelis constants for oxygen and NADPH are 24.9 and 6.5 µM, respectively.
NADP+ is a competitive inhibitor with a
Ki of 1.2 mM. In the absence of oxygenatable substrate
the peroxyflavoprotein intermediate slowly decomposes to the oxidized enzyme
and peroxide [78]. The lipid environment of the monooxygenase
is important to the stability of this intermediate, since phosphatidyl
ethanolamine and phosphatidyl serine effectively diminish uptake of oxygen
in the abscence of substrate [95]. Water, the only other
product of the reaction, is probably released after substrate is oxidized
[96].
Alkylamine and Calcium Effects on the Hepatic Monooxygenase.
Alkylamines and alkylguanidines nearly double the turnovers of substrates
at the pH optimum for the monooxygenase, 8.4 [81,89,97].
At a more physiological pH, 7.4, some preparations are activated 3-fold
[88,97,98].
Although alkylamines and alkylguanidines may prevent inactivation of the
monooxygenase by fatty acids in purified preparations, fatty acids do not
affect activities in microsomes [81]. Degradation of
lipids to free fatty acids should sensitize the monooxygenase to amines,
but activation by alkylamines and alkylguanidines is lost as the enzyme
ages at 4°C. Purification of the monooxygenase on diethylaminoethyl
cellulose, likewise, alters the effect of octylamine on the N-oxygenation
of dimethylaniline (Figure 1).
FIGURE 1
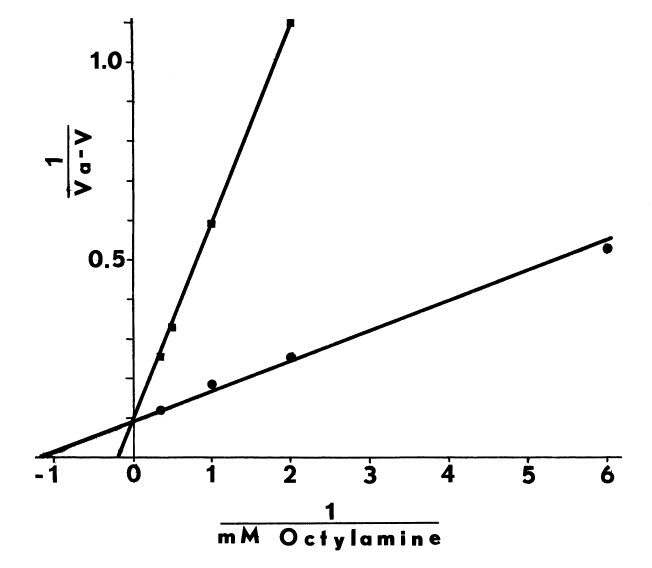
Alkylamines and alkylguanidines, calcium ions
at high concentrations (10 mM), and high pH may all contribute to the destabilization
of the peroxyflavoprotein intermediate, for all three increase substrate-independent
uptake of oxygen. Substrate- independent and substrate-dependent uptake
of oxygen usually doubles as the pH is raised from 7.4 to 8.4 [81,99].
Calcium may sensitize the monooxygenase to changes in pH [99],
since the blank rate triples as the pH is raised over this same range (Figure
2).
FIGURE 2
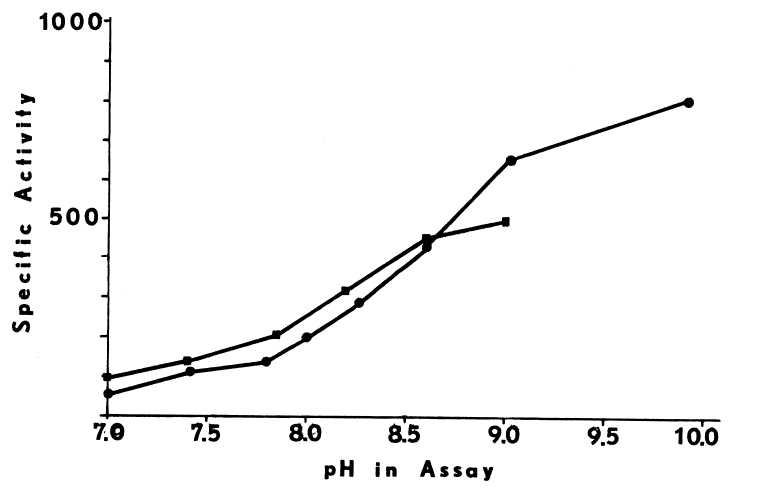
Effects of pH and Temperature on the Monooxygenase. The hepatic monooxygenase is rapidly inactivated at pHs greater than 8.0 and 8.5 when NADH and NADPH, respectively, are supporting substrates [98]. In the abscence of cofactor, the monooxygenase is also extremely heat-labile. NADPH completely prevents irreversible structural changes at 37°C, but at higher temperature (45°C) denaturation is rapid, even in the presence of the cofactor [81,100]. Since insuring the presence of cofactor during purification of the monooxygenase is prohibitively expensive, extremes of temperature (30°C) and pH (8.0) must be avoided at all stages. Indeed, if the liver is not chilled within 10 to 15 minutes after the hog is exsanguinated, the hepatic monooxygenase is inactivated by a rapid post-mortem rise in temperature to 42°C [81,93]. Precautions must be taken even in the microenvironments of various homogenizing and chromatographic techniques.
Reagents. The following reagents are obtained from sources indicated: 5,5'-dithiobis-2-nitrobenzoic acid (DTNB), glucose-6-phosphate, Tricine, NADP+, NAD+, triethylaminoethyl cellulose, coenzyme A, dephosphocoenzyme A, and sephadex from Sigma Chemical Company; pantethine from Calbiochem-Behring Corporation; diethylaminoethyl and carboxymethyl cellulose from Whatman, Ltd.; octanoic acid from California Foundation for Biochemical Research; enzyme-grade sucrose from Mann Research Laboratories; sodium borohydride from MCB Manufacturing Chemists, Inc.; and solvents, 1,4-dithiothreitol, and 2,4-dinitrofluorobenzene (DNFB) from Aldrich Chemical Company. Other purchased reagents are of highest purity available.
Certain reagents are purified prior to use. Cysteamine from Aldrich Chemical Company, n-octylamine from Eastman Chemicals and n-dodecylguanidine from Alfred Bader are recrystallized as the hydrochloride salt from ethanol. N,N-dimethylaniline is purified by preparative gas chromatography. N-methyl-2-mercaptoimidazole (methimazole) from Aldrich Chemical Company is recrystallized from ethyl acetate. Potassium thiocyanate from Allied Chemical is recrystallized from ethanol. Water is glass-distilled from chromic acid.
Other reagents require special handling. Iodide (125I) from Amersham Corporation and potassium iodide are maintained at alkaline pH with potassium hydroxide to prevent nonenzymic oxidation to iodine. Solutions of iodide that have been frozen and isotope that has half-decayed (60 days) are also avoided.
Peptides, Proteins, and Enzymes. Bovine hepatic catalase, glutathione and its reductase, bovine serum albumin, glucose-6- phosphate dehydrogenase, and nucleotide pyrophosphatase (type II from Crotalus adamanteus venom) are obtained from Sigma Chemical Company. Thyroid peroxidase is purified by the procedure of Danner and Morrison [101]. Pepsin is purchased from Worthington Biochemical Corporation. Glutamyl-tyrosyl-glutamate and Z-glutamyl-tyrosine are acquired from Bachem, Inc. and Protein Research Foundation, respectively.
Detergents. Tritons, DN-65 and X-45, are purchased from Sigma
Chemical Corporation. Succinylated Triton X-45, prepared as previously
described, is stored at -20°C to prevent hydrolysis. Storage at lower
temperature also retards potentially harmful peroxidation [93].
Deoxycholate is conveniently stored at room temperature, since it is used
to disperse protein in the biuret assay.
Peptides for Iodination. Half-cystinyl P-9 peptide is isolated from bovine serum albumin by a procedure derived from those used in the purification of P-9 [102] and KL [103] fragments. Twenty grams of bovine serum albumin are dissolved in 280 mls of 0.12 M ammonium formate buffer containing 0.42 g cystine. After adjusting the pH to 8.0, the mixture is allowed to stir overnight at room temperature. Octanoic acid (0.4 ml) is then added and the pH is lowered to 3.7 with 0.12 M formic acid. A fresh solution of pepsin (20 mg in 40 ml of 1 mM HCl) is then added and incubation proceeds with stirring for 20 minutes at 25°C. Hydrolysis is halted by raising the pH to 7.4 with 2 M ammonium hydroxide. After stirring for one hour, the digest is chilled in ice and is treated with sufficient cold 5 M TCA to lower the pH to 4.2. The pellet resulting from a 1200xg centrifugation at 4°C is resuspended in a minimum volume of ammonium formate buffer. The pH is then adjusted to 5.2 and peptides are precipitated with 20 volumes of acetone. Small quantities of magnesium chloride facilitate precipitation. The resulting pellet is washed three times by resuspending and precipitating in acetone. The final pellet is air-dried to a powder over a warming plate. The crude P-9 extract is then dissolved in a minumum volume of 0.23 M ammonium formate buffer, adjusted to pH 3.5, and chromatographed on a 2.5 by 60 cm, sephadex G-5O column equilibrated with the same buffer. The volume of sample does not exceed 2 percent of the column's bed volume. Retained fractions containing the greatest ratio of absorbance at 280 nm to protein (biuret) are pooled. Ammonium formate is removed by precipitating the neutralized peptide with spectral grade acetone. After drying, the peptide is resuspended in 10 mM potassium phosphate buffer (pH 7.5) and frozen in aliquots at -20°C for use as substrate for iodination. Electrophoresis of the P-9 peptide displays a single band migrating between cytochrome c and a trailing fraction from the sephadex column. The trailing fraction exhibits a lower ratio of absorbance at 280 nm to protein and probably contains P-6 and P-Phe fragments. Concentrations for peptidyl iodination are based upon an estimated millimolar extinction coefficient at 274.5 nm of 10 for the pure half-cystinyl P-9 fragment.
Lysylglutaryl P-9 is prepared by sequentially reacting 0.25 M glutaraldehyde and lysine in excess with purified half-cystinyl P-9 peptide at pH 6.0. The Schiff bases are reduced with sodium borohydride to stabilize the blocking group. Chromatography on sephadex G-50 removes unreacted reagents.
N-Acetyl-glutamyl-tyrosyl-glutamate is prepared from the tripeptide by acetylating with acetic anhydride and deacylating the tyrosyl residue with hydroxylamine [104]. Glutamyl-tyrosyl-glutamate (250 mg) is dissolved in a minimum volume of saturated sodium acetate.
The mixture is chilled in an ice bath, and
the pH is adjusted to 8.5 with 6 N sodium hydroxide. Ten µliters
of acetic anhydride are then added every two to three minutes for one hour.
Between injections of the anhydride, the pH is adjusted with 6 N sodium
hydroxide to maintain the pH between 7.5 and 9.0. When the pH has stabilized
after the last injection, an excess of hydroxylamine is added. The deacylation
is allowed to proceed overnight at 0ºC. The crude product is chromatographically
desalted with sephadex G-10. Fractions absorbing at 274.5 nm are pooled
and lyophilized. The coarse powder is dissolved in a minimum volume of
water and the pH is lowered to 3.0 with phosphoric acid. Extraction with
three twenty-volume aliquots of acetone removes colored material. The pH
is adjusted to 6.0 with potassium hydroxide and the aqueous phase is again
extracted with acetone. If necessary, this phase is lyophilized to a powder.
Three extracts of glacial acetic acid (ten parts by weight of powder) are
pooled and treated with twenty volumes of acetone. The precipitate from
acetone is dried in vacuo, resuspended in water, and adjusted to pH 7.4
with potassium hydroxide. Concentrations of N-acetyl-glutamyl-tyrosyl-glutamate
are estimated based upon the millimolar extinction coefficient of tyrosine
at 274.5 nm of 1.4 [23]. These estimates may be exaggerated
since the average for tyrosyl residues within the half-cystinyl P-9 peptide
is 1.68 liter/mmole/cm.
Preparation of Diethylaminoethyl Cellulose Acetate. Fresh diethylaminoethyl cellulose is washed first with ten volumes of glass-distilled water and, then, with ten volumes of potassium phosphate buffer (pH 7.0). A slurry of the washed material in phosphate buffer is then lowered to pH 7.0 with acetic acid before the column is poured. Preparation in this manner helps to prevent alkaline denaturation of the monooxygenase on the column.
Preparation of Crude 4-Phosphopantetheine. Three µmoles of dephosphocoenzyme A is hydrolyzed to 4-phosphopantetheine and AMP by nucleotide pyrophosphatase. The 1 ml hydrolysis mixture contains 10 mM magnesium chloride and is buffered at pH 7.4 with 10 mM Tris-(hydroxymethyl)-aminomethane Sufficient pyrophosphatase is added to hydrolyze 3 µmoles of NAD+ per minute. The reaction is allowed to proceed at 25°C for 20 minutes. The hydrolysate is then chilled on ice and chromatographed over sephadex G-10 to remove the pyrophosphatase. As the column is eluted with 10 mM potassium phosphate buffer (pH 7.4), the effluent is monitored at 259 nm for adenylic acid. 4-phosphopantetheine, having a similar molecular weight, co-elutes.
Miscellaneous. Ultrafiltration membranes are obtained from Amicon Corporation.
Assay of Glutathione and Cysteamine. Glutathione is estimated as cytosolic, acid-soluble thiol with Ellman's reagent [105]. Thiol to disulfide ratios are also determined using DTNB. Cysteamine is determined using DNFB. [106].
Assays of Enzymes Influencing Thiol to Disulfide Ratios. Glutathione reductase [107], glutathione peroxidase [108] and thioltransferase [109] in the cytosolic fraction are assayed as described except for pH (7.4) and temperature (37°C). The combined action of these enzymes creates a reductive environment in the cytosol by (1) removing peroxide and (2) reducing disulfides (RSSR) in the following reactions:
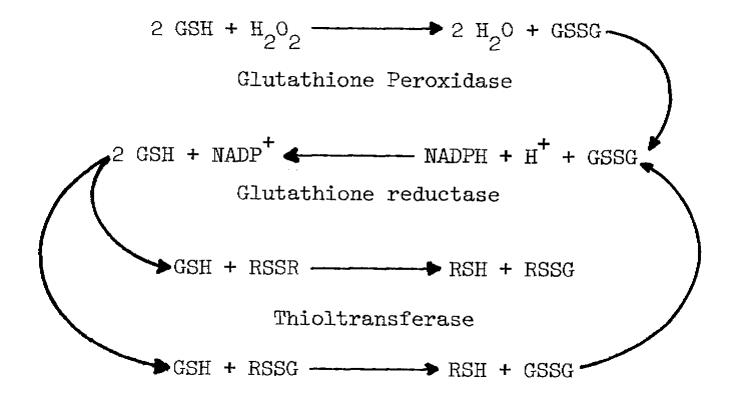
Catalase also catalyzes the removal of peroxide [110]. Production of oxygen is monitored polarographically [78] in deoxygenated (boiled) potassium phosphate buffer (pH 7.4) at several concentrations of peroxide.
Assay of Microsomal FAD-Containing Monooxygenase. Understanding oxidation of iodide catalyzed by the hepatic monooxygenase is an important step in assessing the role of this enzyme in the biosynthesis of thyroid hormones. Although enzymic activity can be monitored by following changes in any of the substrates or products, it is more convenient to follow uptake of oxygen [78] when multiple substrates and products are involved. The substrate-independent rate of oxygen uptake increases as the monooxygenase is purified. NADPH and oxygen are consumed in the blank rate, but little intermediary iodine is produced in the presence of catalase. Most preparations of the monooxygenase are contaminated with a twenty-fold catalytic excess of catalase. Since lipias also influence blank rates, monooxygenase preparations exhibiting little substrate-independent uptake of oxygen are selected for the following studies. Problems arising from the generation of peroxide are diminished further by assaying at pH 7.4 (Figure 2).
Another unavoidable aspect of these studies is that the high concentrations of iodide necessary to obtain accurate kinetic values for the monooxygenase also lead to irreversible oxidation of the supporting substrate, NADPH. Low concentrations of iodide are employed when possible to circumvent this problem.
Although hepatic monooxygenase is assayed polarographically in aerated buffer [78], this method is not sufficiently sensitive to detect monooxygenase in thyroidal preparations. N,N-dimethylaniline N-oxygenation [111] semi-quantitatively indicates monooxygenase in crude preparations. The most sensitive assay for the monooxygenase, however, utilizes Ellman's reagent [112]. DTNB (10 mM) is reduced with stoichiometric amounts of sodium borohydride, and allowed to stand overnight at 4ºC to ensure complete destruction of the borohydride. Sufficient reduced DTNB is added to each ml of media to obtain an absorbance of one at 412 nm. The assays are buffered at pH 8.4 with 0.1 M Tricine and contain 0.6 mM NADP+, 2.5 mM glucose-6-phosphate, and one IU of glucose-6-phosphate dehydrogenase.
After equilibrating at 37°C for 5 minutes, a sample containing monooxygenase is added, mixed, and monitored at 412 nm in a Gilford recording spectrophotometer. Since reduced DTNB is not a substrate for the monooxygenase, oxidation at this stage reflects substrate-independent reactions. Adding methimazole (1 mM) establishes a substrate-dependent rate. The sulfenic acid of methimazole at this pH nonenzymically oxidizes two moles of reduced DTNB, thereby regenerating methimazole. Rates are calculated by subtracting the substrate-independent rate, dividing by two, and dividing by the millimolar extinction coefficient of reduced DTNB (13.6 liter/mmole/cm) [105].
Like other reactions dependent upon the hepatic monooxygenase, the rate of oxidation of reduced DTNB is doubled by alkylamines and alkylguanidines. Unfortunately, this method is considerably more prone to interference. Any factor influencing NADPH-dependent reduction of DTNB [113] or glutathione dramatically diminishes the oxidation of reduced DTNB. Similarly, generation of oxidants by other mechanisms, such as the peroxidation of lipids and detergents, contributes to spurious rates. Since turbidity, pyrophosphate, EDTA, contaminating enzymes, detergent, and glutathione are all problematic, analysis is most accurate with purified preparations and under tightly controlled conditions.
Assay of Glucose Oxidase. Increasing glucose to 0.5 M nearly saturates glucose oxidase [114], making the rate of peroxide generation dependent upon added oxidase. Consumption of oxygen catalyzed by glucose oxidase is assayed polarographically [78] under conditions identical to the peroxidase assays.
Assay of Thyroid Peroxidase. Generation of periodide and iodination
of peptides are followed spectrophotometrically at 37ºC. Molar extinction
coefficients are 22,900 at 353 nm for periodide [115]
and 2430 at 290 nm for glutamyl-iodotyrosyl-glutamate [49].
Either procedure uses 0.1 M potassium phosphate buffer (pH 7.4), 0.5 M
glucose, and varying amounts of glucose oxidase [64].
One-half or ten mM iodide is used in assaying for peroxide-dependent iodination
and peroxide-dependent generation of periodide, respectively [48].
Glucose oxidase is added last.
Assay of NADPH-Dependent Iodination. NADPH-dependent iodination
is measured by determining the rate of incorporation of isotope into the
half-cystinyl P-9 peptide. Total volume is limited to 0.4 ml in a 1.5 by
12.5 cm, thick-walled test tube to ensure adequate aeration. The media
contain sequentially as added 0.1M potassium phosphate buffer (pH 7.4),
0.6 mM NADP+, 4 mM glucose-6-phosphate,
4 IU of glucose-6-phosphate dehydrogenase, 0.5 mM glutathione, 10 µM
potassium iodide, 50 µM half-cystinyl P-9, 25 µM cysteamine,
and 50 nCi of 125I. Adding glutathione
and unlabelled iodide to the assaying media before cysteamine and the isotope
prevents nonenzymic formation of product. The mixture is preincubated at
37ºC for two minutes in an agitating water bath to ensure protective
concentrations of reduced pyridine nucleotide. Samples are then injected
and incubation proceeds with agitation for, at most, 3 minutes. Iodination
is terminated with 20 µl of 2 M sodium bisulfite [115]
and chilling on ice. Magnesium chloride (5 µ1 of a 1 M solution)
and 20 volumes of acetone are immediately added to each tube. The tubes
are then carefully mixed on a vortexing mixer, and centrifuged for five
minutes at high speed in a clinical centrifuge. The supernatant contains
unbound tracer and is decanted for later concentration, in vacuo, and disposal.
The pellet is dispersed in 2 ml of acetone with a stirring rod by vortexing
the tube. The powdery suspension is then centrifuged as before for one
minute. After decanting the supernatant, the pellet is washed twice more
by dispersing and centrifuging in 2-ml aliquots of acetone. The resulting
pellet is resuspended in distilled water and counted for one hour in a
Beckman Gamma 4000 counter. Data is corrected for background, nonenzymic
iodination, radioactive decay, and dilution of isotope by unlabelled iodide.
Isotopic effects and quenching are presumed inconsequential in these initial
studies.
Assay of Peroxide-Dependent Iodination. Peroxide-dependent iodination is monitored in essentially the same manner as NADPH-dependent iodination. Glucose (0.5 M) and varying amounts of glucose oxidase replace NADP+, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase as generating system in the media. Glutathione is also excluded from the assaying mixture except where noted.
Assay for Iodination with Small Peptides. Smaller peptides, glutamyl-tyrosyl-glutamate
and its N-acetylated derivative, are more soluble in acetone. To circumvent
problems in recovering product, iodine not bound to tyrosyl residues is
removed by a different procedure. Iodination, performed as with half-cystinyl
P-9, is terminated with 20 volumes of acetone saturated in sodium bisulfite.
The medium is transferred to a separate column consisting of triethylaminoethyl
cellulose in a polypropylene, pipetting tip. The fluid is aspirated through,
and the column is then washed with acetone containing 10 mM potassium iodide
and saturated in sodium sulfite. When isotope no longer elutes from the
column (5 to 10 mls), the entire column is then placed in a vial and counted
as before. Again no corrections are made for quenching.
Slab-gel Electrophoresis. Peptides and proteins are resolved on polyacrylamide gels containing sodium dodecyl sulfate and 1.5 percent 1,4-dithiothreitol [102,116]. Coomassie brilliant blue R is used to visualize protein. Integral membrane proteins such as the monooxygenase [81] bind inordinate amounts of detergent, so mobilities tend to be unusually high. Doubling the detergent in gels and running buffers ensures saturation in sodium dodecyl sulfate and, ultimately, resolution of these hydrophobic proteins and peptides.
Protein Determination. Protein concentrations are estimated by the biuret method [117]. Sodium deoxycholate, 0.3 µg/ml, is routinely added to disperse lipids and resuspend precipitated proteins. Occasionally, turbidity remains even after treatment with deoxycholate. Extraction with an equal volume of ethyl ether followed by centrifugation in a clinical centrifuge clarifies the lower layer. Other special precautions must be taken when heme is present, since absorbance of this chromophore at 540 nm leads to overestimation of protein. Precipitation of the protein with 0.3 M trichioroacetic acid eliminates the heme-dependent absorbance.
Preparation of Heme Derivatives. Pyridine hemochromogen, deoxy-, and carbonmonoxy-derivatives are prepared essentially as described for hemoglobulin [118]. Spectra are obtained with an Aminco DW-2aTM spectrophotometer.
Assay of Cytochrome c Reductase. Cytochrome c reductase is assayed as previously described [54].
TISSUE COLLECTION AND SUBCELLULAR FRACTIONATION
Extreme measures must be taken in the collection of tissue to ensure microsomal NADPH-dependet iodinating activity. The thyroid is unavoidably exposed to adverse conditions at the abattoir. Hogs are stunned by cerebral puncture, hung by the hind-quarters, and exsanguinated by puncturing the superior vena cava. Although this incision makes the thyroid readily accessible, the tissue is bathed in hot deoxygenated blood. To minimize denaturation of activities within, the thyroid is removed immediately after the incision and chilled on ice in cold 0.25 M sucrose. The elapsed time from incising until chilling is usually less than 15 seconds. Estimated elapsed time from stunning until chilling is three minutes. Thermal inactivation as a function of the latter elapsed time is illustrated by the following table of microsomal activities:

Preparation of Microsomes. Thyroidal microsomes are purified by a procedure similar to one used in the preparation of hepatic microsomes [93]. The thyroids are quickly stripped of connective tissue and fat upon arrival from the slaughterhouse and stored in cold (4°C) sucrose maintained at pH 7.7 until they are processed. All subsequent processing and purification procedures are performed at 4°C.
Thyroids (0.5 kg) are weighed, minced in a
hand grinder, and suspended in 3 volumes of 0.25 M sucrose. After adjusting
the pH to 7.7, the slurry is passed through a continuous flow homogenizer
[119].
The homogenate (Figure 3, Table 1)
is then adjusted to pH 7.7 with 1 M potassium hydroxide and centrifuged
for one hour in an International B-35 centrifuge at 10,000 rpm (12,000xg).
The sedimentnuclei, mitochondria, and stromacontains less than 25 percent
of the original iodinating activity. The post-mitochondrial supernatant
is decanted through cheesecloth to remove lipid and then adjusted to pH
5.7 with 1 N acetic acid. Centrifugation for one hour at 30,000 rpm in
a Beckman Model L Ultracentrifuge sediments microsomes. The crude pellet
is washed twice with equal volumes of wash buffer, 1 mM glutathione buffered
at pH 7.8 with 10 mM potassium phosphate. This is done by resuspending
in wash buffer and carefully raising the pH to 7.7 with 1 M potassium hydroxide.
After homogenizing in a teflon-glass homogenizer, the pH is carefully lowered
to 5.7 with 1 M acetic acid. Centrifuging as before at 30,000 rpm completes
one wash cycle.
FIGURE 3
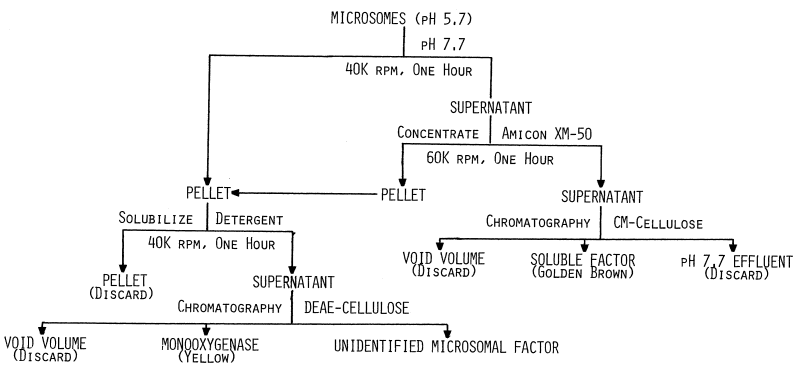
Flow chart for resolving and purifying components of the thyroidal iodinating system. Centrifugation is represented by speed in thousands of revolutions per minute (K rpm). All procedures are carried out at 4°C.
The acidic, post-microsomal supernatants (all
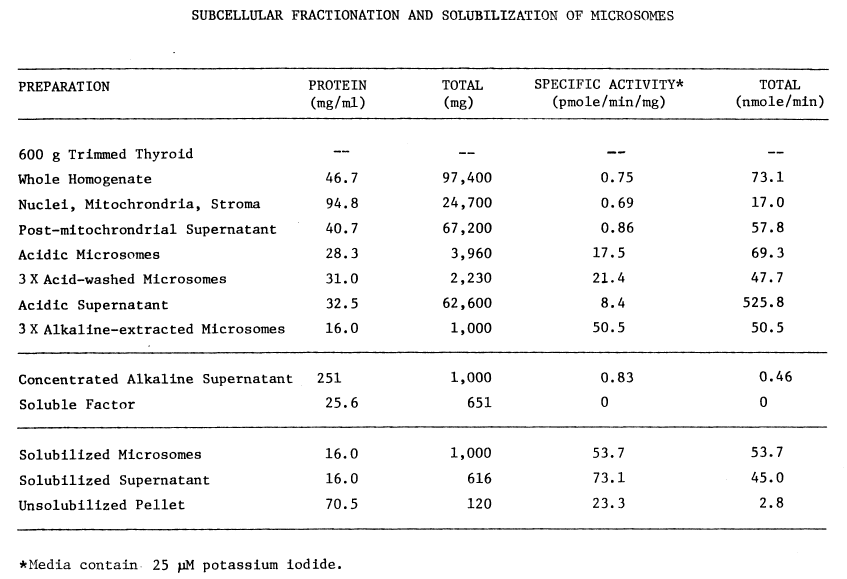
three) contain appreciable iodinating activity (Table 1),
but are contaminated with soluble proteins. The washed, microsomal pellet
contains at least 60 percent of the original activity. Since washing the
microsomes at pH 7.7 or with 50 µM pyrophosphate causes losses of
iodinating activity, these treatments are avoided in the purification of
microsomes. After resuspending in an equal volume of wash buffer at pH
7.0 and containing 0.25 M sucrose, the microsomes can be frozen at -20°C
with little loss of activity.
TABLE 1
ENDOGENOUS MODULATORS OF THE IODINATING SYSTEM
The post-particulate supernatant contains all
of the components contributing to a reducing environment: acidsoluble
thiol (glutathione), glutathione reductase, catalase, thioltransferase,
and glutathione peroxidase (Table 2). Even so, the average
thiol to disulfide ratio in protein from three thyroids is quite low, 11.1
percent. Total thiol (66 µM), including reduced proteinaceous disulfides
and mixed disulfides, is nearly an order of magnitude less than the concentration
of acid-soluble thiol 416 µM. Free thiol appears much more abundant
than disulfide, the latter being localized in protein.
TABLE 2
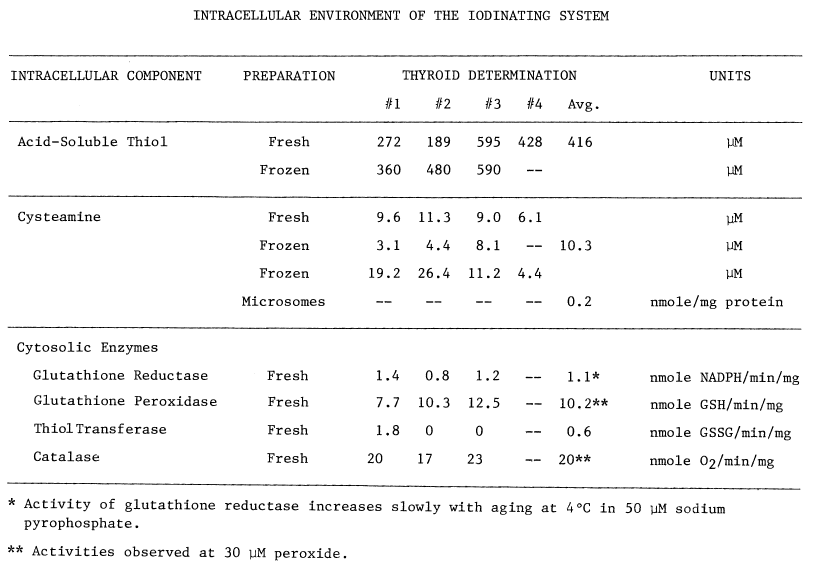
Cysteamine is also present (Table
2), and with the monooxygenase and thioltransferase activities constitutes
an oxidizing system. An average of at least 10 µM cysteamine exists
in the thyroid. A considerable amount of cysteamine is also sporadically
observed in microsomes. Washing at pH 5.7 and then at 7.7 lowers the concentration
of cysteamine in microsomes, but detectable amounts often remain.
VERIFICATION OF ASSAY FOR THYROIDAL NADPH-DEPENDENT IODINATION
A fundamental problem in establishing an NADPH-dependent
iodinating system is finding a suitable acceptor for oxidized iodide. Of
the substances testedtyrosine, Z-glutamyl-tyrosine, glutamyl-tyrosyl-glutamate,
N-acetyl-glutamyl-tyrosyl-glutamate, half-cystinyl P-9, and thyroglobulinonly
three are appreciably iodinated by the microsomal NADPH-dependent system
(Figures 4,5). Thyroglobulin is rapidly
deiodinated (unreported observation), is somewhat variable in composition
[23],
and is not readily precipitated with acetone. N-acetyl-glutamyl-tyrosyl-glutamate,
though chemically well-defined, becomes increasingly inhibitory as microsomes
age (Figure 5). While the trend in activity and apparent
Km can be partially reversed, substrate inhibition
is unaffected by 0.1 M potassium chloride. Magnesium acetate (10 mM) is
without effect, but further addition of as little as 10 µM calcium
acetate reverses most of the peptide-dependent inhibition.
FIGURE 4
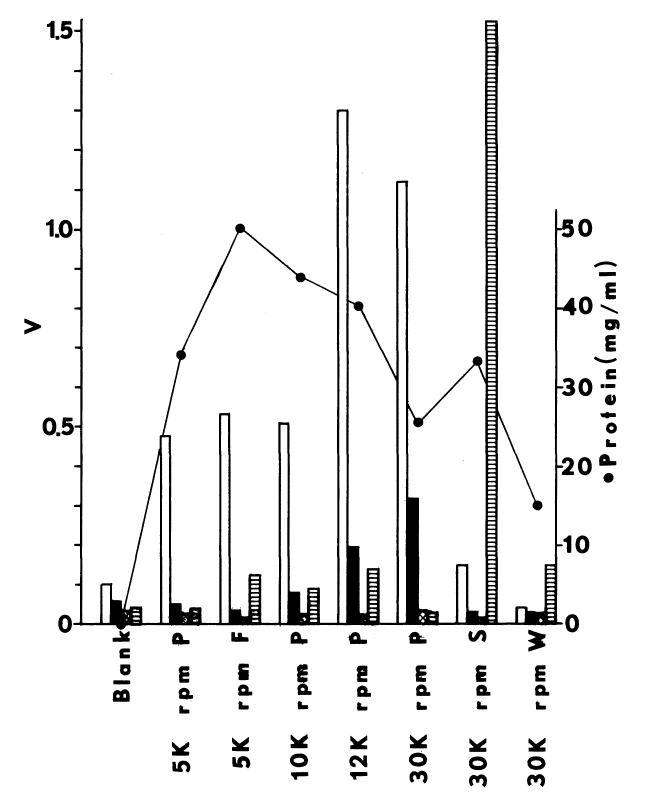
Subcellular distribution and specificity of thyroidal iodinating activities. Subcellular fractions are obtained by differential one-hour centrifugations at alkaline pH. Iodinating velocities (µM/minute) for each fraction are represented by bars coded for assaying conditions. NADPH-dependent iodination of half-cystinyl P-9 or glutamyl-tyrosyl-glutamate is depicted by open and striped bars, respectively. Concentration of cysteamine in the media is double that present in the standard medium described in METHODS. Under similar conditions very little peroxide-dependent iodination of half-cystinyl P-9 is observed (crosshatched bars).
Solid bars portray peroxide-dependent iodination
of glutamyl-tyrosyl-glutamate in the abscence of glutathione and cysteamine.
Rate of peroxide generation in these media is 50 µM per minute. All
media contain 250 µM iodide to compare NADPH- and peroxide-dependent
iodination under conditions favoring the latter, Circles indicate protein
estimates of the various subcellular fractions. Fractions assayed from
left to right are as follows: nuclei, mitochondria, and stroma (5K rpm
P); a fluffy intermediary layer (5K rpm F); a heavy particulate pellet
(10K rpm P); a heavy microsomal pellet (12K rpm P); a light microsomal
pellet (30K rpm P); the post-microsomal supernatant (30K rpm S); and a
supernatant obtained by resuspending and resedimenting microsomes (30K
rpm W).
FIGURE 5
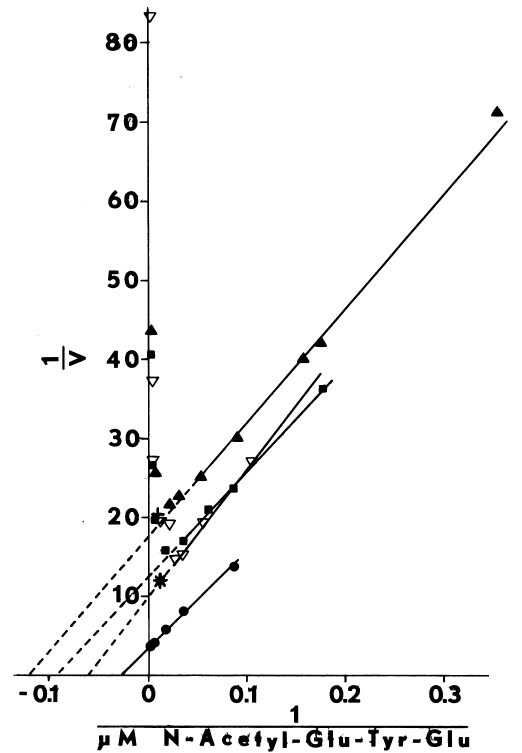
Effect of aging upon NADPH-dependent iodination. Iodination of
N-acetyl-glutamyl-tyrosyl-glutamate is determined as described in METHODS,
except the concentration of iodide in the media is increased to 250 µM.
Velocities (V) are expressed as nmole/minute/mg microsomal protein. Microsomes
are assayed the day they are prepared (circle), and after two (square),
and four (filled triangle) days of aging at 4°C. Adding 10 mM magnesium
acetate (+) does not influence iodination catalyzed by aged microsomes.
Supplementing with magnesium acetate and 10 µM calcium acetate (*)
restores much of the activity observed prior to aging. Some effects of
aging are also ameliorated by 0.1 M potassium chloride (inverted triangle)
at low concentrations of the tripeptide.
Iodination of Half-Cystinyl P-9. Half-cystinyl P-9 also contains a glutamyl-tyrosyl-glutamyl sequence [120] and is inhibitory in the microsomal system. This peptide, nonetheless, has several redeeming characteristics. Since it is easily precipitated and separated from unbound iodide by washing in acetone, product can be isolated at neutral pH, thereby avoiding isotopic exchange that reportedly occurs at the more acidic pH [115]. Analysis of material recovered in the acetone-washed pellet on sephadex G-200 indicates that half-cystinyl P-9 is exclusively iodinated. Without added peptide a heterogeneous group of microsomal proteins is iodinated. This peptide, therefore, provides an accurate representation of iodinating activity during purification.
NADPH-dependent iodination of peptide is presented in Figure 6. Linearity is observed for three minutes. Sodium bisulfite disrupts association of iodine with amino acids such as lysine and histidine, but does not influence the more covalent bonding in iodotyrosyl residues [115]. Apparently, one-fourth of the iodine is bound to tyrosyl residues in media lacking amines. The diamine, cystamine, does not influence overall iodinating activity, but linearity with time is improved (Figure 6). NADPH-dependent iodination is, ultimately, more specific for tyrosyl residues when cystamine is included; up to 60 percent is insensitive to sodium bisulfite (Figure 7). Other substrates for the hepatic monooxygenase are potent inhibitors of NADPH-dependent iodination (Table 3). Inhibition is essentially complete, except at high concentrations of cystamine. Dodecylguanidine, an activator for the hepatic monooxygenase, slows the overall iodinating rate and nearly eliminates bisulfite-insensitive iodination (Figure 6).
Peroxide-dependent iodination, in contrast
to that dependent upon NADPH, is slower and quite labile under these more
physiological conditions (Figure 7); linearity with time
can only be obtained at rates of iodination fractional to NADPH-dependent
rates. Attempts to increase the rate to NADPH-dependent levels by increasing
generation of peroxide result in prompt inactivation of the peroxidase.
FIGURE 6
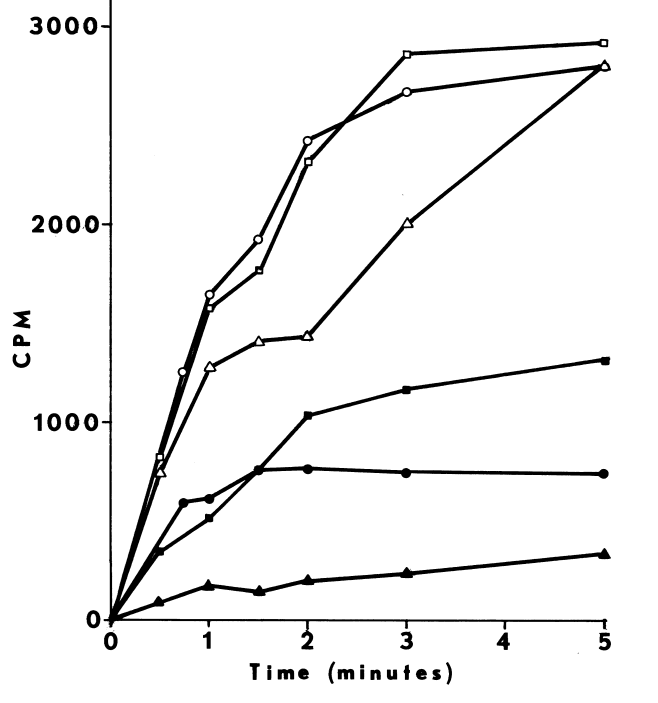
Effect of cystamine and dodecylguanidine on the specificity of NADPH-dependent
iodination. Iodination of half-cystinyl P-9 catalyzed by thyroidal
microsomes is determined as described in METHODS, except glutathione and
cysteamine are excluded. Open symbols represent counts/minute/assay (CPM)
incorporated upon assaying with the following supplements: none (circle);
25 µM cystamine (square); and 0.3 mM dodecyl guanidine (triangle).
Closed symbols depict counts/minute/assay (CPM) remaining after treating
with molar sodium bisulfite and washing with acetone.
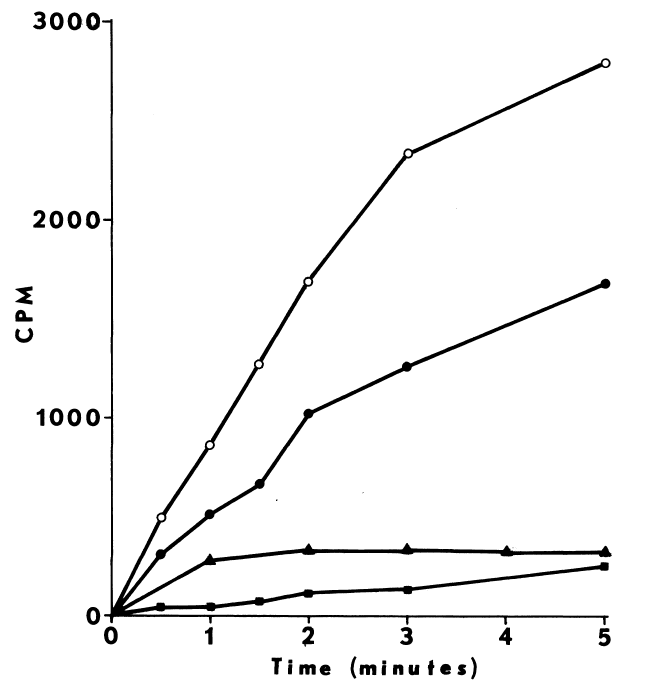
Comparison of peroxide- and NADPH-dependent iodination. Iodination
of half-cystinyl P-9 catalyzed by thyroidal microsomes is determined as
described in METHODS with the following changes: glutathione is excluded,
and cystamine replaces cysteamine in the media. Open symbols depict total
counts/minute/assay (CPM) incorporated, while closed symbols indicate radioactivity
remaining after treating with molar sodium bisulfite and washing in acetone.
NADPH-dependent iodination is represented by circles (open,filled). Peroxide-dependent
iodination is determined at two rates of peroxide generation: 15 µM
H202 /minute (square) and 93 µM H202/minute
(triangle).
TABLE 3
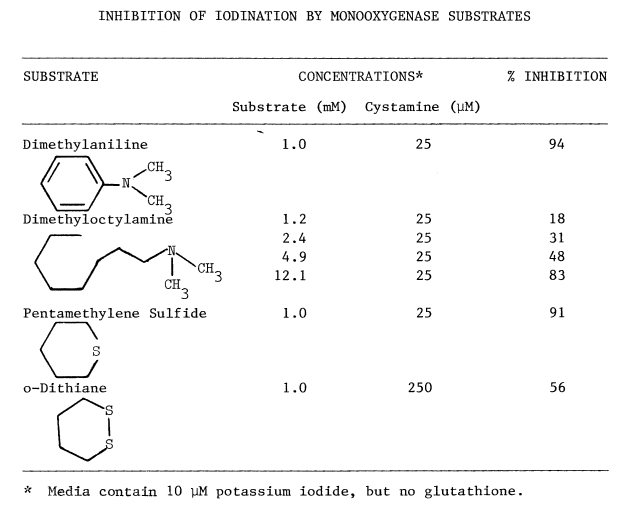
Elimination of Peroxide-Dependent Iodination. Low concentrations
of the endogenous reductant, glutathione, when added to the NADPH-dependent
system have no appreciable effect on bisulfite-insensitive jodination.
Peroxide-dependent iodination, in contrast, is greatly diminished even
at the more favorable, but less physiological, concentration of iodide
(Table 4).
TABLE 4

Overall efficiency of peroxide-dependent iodination is affected also. Even though generation of peroxide by glucose oxidase (34 nmole/min/mg microsomal protein) is two orders of magnitude greater than monooxygenase activity (0.29 nmole/min/mg protein), NADPH-dependent rates are 3 times faster than peroxide-dependent iodination in the presence of glutathione (Table 4). The uptake of oxygen catalyzed by glucose oxidase is unaffected by glutathione.
Optimal Concentrations of Glutathione and Cysteamine. Cysteamine,
like cystamine, stimulates iodination catalyzed by thyroidal microsomes.
Iodination is enhanced at concentrations less than 40 µM, and optimal
at 10 to 20 µM (Figure 8).
FIGURE 8
Stimulation by glutathione is evident as well,
but at much higher concentrations, 0.5 to 1 mM (Figure 9).
Raising glutathione further inhibits but does not return activity to untreated
levels. Iodination catalyzed by microsomes that have aged for 4 days at
4°C is more sensitive to glutathione. During this time as much as 60
percent of the original activity is lost. The remaining activity is inhibited
by 4 to 6 mM glutathione.
FIGURE 9
Effect of glutathione on NADPH-dependent iodination. Iodination
of half-cystinyl P-9 catalyzed by thyroidal microsomes is determined as
described in METHODS with the following changes: the concentration of cysteamine
is diminished to 15.8 µM, and glutathione is varied over the range
of concentrations indicated (filled circles). Velocities (V) are expressed
as nmole/minute/mg microsomal protein. Iodination catalyzed by microsomes
that have aged 6 days at 4°C is only 40 percent of the activity present
in fresh microsomes. Correcting for this loss illustrates a disproportionate
suppression of activity in aged microsomes at 4 to 6 mM glutathione (open
circles).
Differentiation with Thiocyanate. Low concentrations of thiocyanate,
20 to 100 µM, can be used to distinguish peroxide-dependent iodination
from that which is NADPH-dependent (Figure 10). At 60
µM thiocyanate, the former activity can be augmented 2-fold, depending
upon the availability of peroxide. NADPH-dependent iodination, in contrast,
is half-inhibited by 83 µM thiocyanate (Figure 11).
Uptake of oxygen catalyzed by the hepatic monooxygenase is significantly
inhibited only at concentrations of thiocyanate greater than 1 mM.
FIGURE 10
Differentiation of peroxide- and NADPH-dependent iodination with
thiocyanate. Iodination of half-cystinyl P-9 catalyzed by thyroidal
microsomes is determined as described in METHODS. The media contain 1.4
mg of microsomal protein, 0.25 mM cystamine instead of cysteamine, and
no glutathione. NADPH-dependent iodination (circle) is inhibited by thiocyanate.
Peroxide-dependent iodination is initially stimulated. Stimulation is greater
when peroxide is generated at 285 µM H202/minute
(filled triangle) than at 16 µM H202/minute
(open triangle).
FIGURE 11
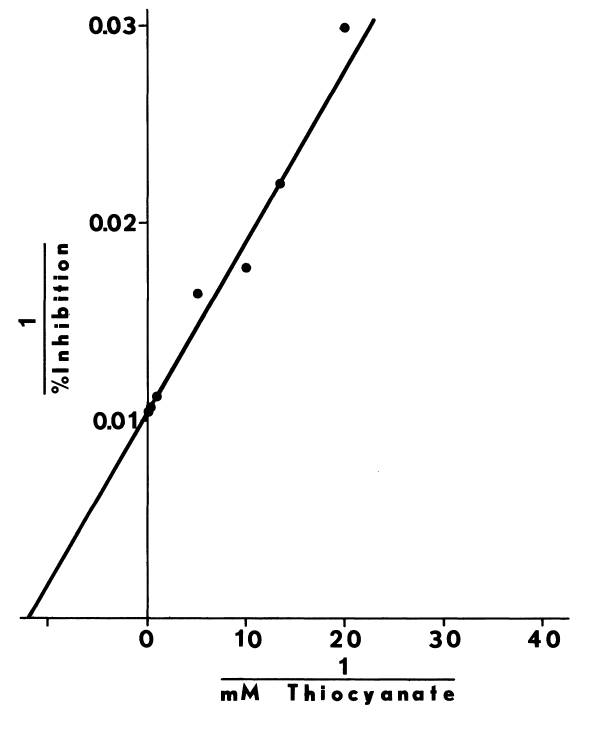
Inhibition of NADPH-dependent iodination by thiocyanate. NADPH-dependent
iodination of half-cystinyl P-9 catalyzed by thyroidal microsomes is determined
as described in the legend of Figure 10. Percent inhibition
in this double reciprocal plot represents the fraction of iodinating activity
lost at each concentration of thiocyanate. Inhibition is virtually complete
at mmolar thiocyanate.
RESOLUTION OF COMPONENTS
Although thyroid peroxidase, monooxygenase, and NADPH-dependent
iodinating activities can be solubilized from thyroidal microsomes, purification
and resolution of the solubilized complex into components is quite difficult;
a fraction purified 2,900 fold contains only 2 percent of the activity
present in the homogenate. Partially resolving the system, prior to solubilization
with detergents, averts problems in both recovery and resolution (Table
1). One component, the soluble factor, precipitates with microsomes
at pH 5.7. Most of the contaminating proteins remain in the acidic supernatant,
as the low specific activity indicates. An endogenous inhibitor also precipitates
at acidic pH, since total activity in the supernatant far exceeds that
present in the homogenate. Soluble factor is extracted from the microsomes
at pH 7.7. Freezing facilitates removal of both endogenous inhibitor and
soluble factor from microsomal preparations (Table 5).
Solubilization of integral membraneous components is also enhanced.
TABLE 5
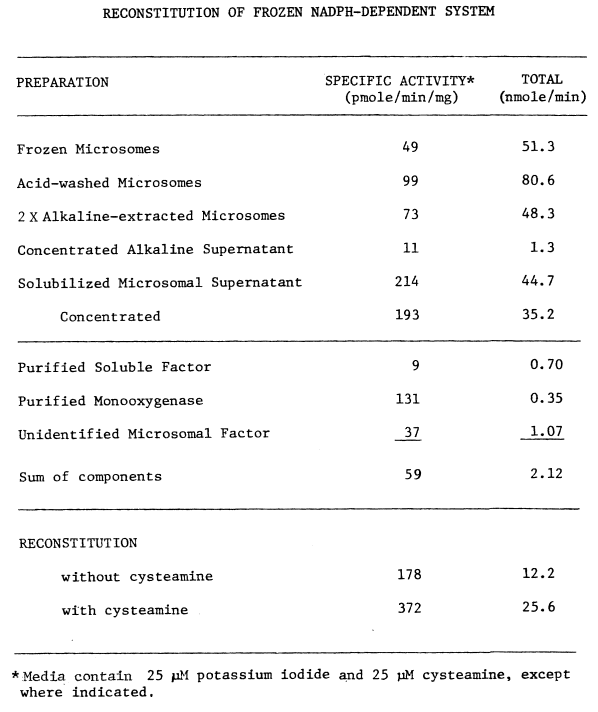
Purification of Soluble Factor. (Figure 3).
Fresh microsomes are resuspended in an equal volume of water buffered at
pH 7.7 with 10 mM potassium phosphate. These microsomes, or frozen ones
that have been thawed slowly on ice (Table 5), are then
adjusted to 7.7 with 1 M potassium hydroxide. After homogenizing in a teflon-glass
homogenizer, the microsomes are centrifuged for one hour at 40,000 rpm.
This represents one extraction cycle. The supernatant is removed and saved,
while the pellet is extracted twice more by resuspending, homogenizing,
and sedimenting. The final pellet, alkaline-washed microsomes, can be
stored frozen by resuspending in glutathione, sucrose, and buffer. The
supernatants are concentrated to 4 ml under argon using an Amicon XM-50
ultrafiltration membrane. Centrifugation at 60,000 rpm in a Beckman L265B
centrifuge for 1 hour removes suspended microsomes. After lowering the
pH to 6.5, the supernatant is chromatographed with 10 mM potassium phosphate
buffer (pH 7.0) on a 2.5 x 30 cm carboxymethyl cellulose column equilibrated
with the same buffer (Figure 12). A golden to reddish-brown
fraction trailing an initially yellow effluent is the soluble factor. The
heme to protein ratios in the three peak fractions are 14.3, 16.6, and
18.5 nmole/mg protein. Lacking detectable peroxidase activity (Tables 1,5),
the soluble factor can be assayed qualitatively in the NADPH-dependent
system. The purified protein is stored in glutathione, sucrose, and potassium
phosphate buffer at -20°C. Repeated freezing and thawing decreases
absorbance at 413 nm.
FIGURE 12
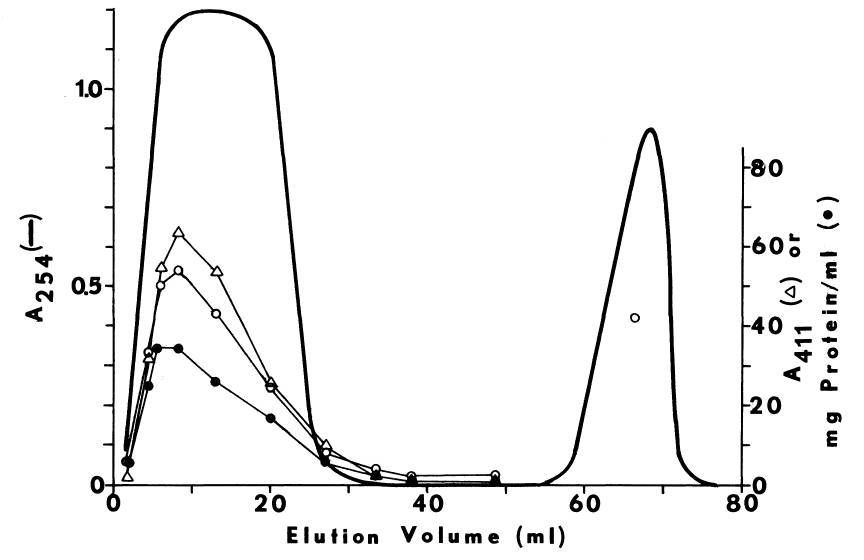
Purification of Microsomal FAD-Containing Monooxygenase. Thyroidal
flavin-containing monooxygenase is purified by a procedure (Figures 3,13)
developed to quickly purify the solubilized hepatic enzyme. Alkaline-extracted
thyroidal microsomes are diluted to 20 mg protein/ml and solubilized with
1.2 mg detergent/mg protein at pH 7.7. The detergent contains 4 parts Triton
DN-65 and one part succinyl-Triton X-45. After homogenizing in a glass-teflon
homogenizer and stirring for 30 minutes, the solubilized microsomes are
centrifuged for one hour at 40,000 rpm. The supernatant is concentrated
to 4 ml using the XM-50 ultrafiltration membrane. The crude, detergent
extract is lowered to pH 6.2 with 1 M acetic acid and chromatographed on
diethylaminoethyl cellulose acetate, prepared as described in METHODS.
Material eluting with 10 mM potassium phosphate buffer (pH 7.0) is assayed
for activity, but usually discarded. Monooxygenase and soluble factor usually
bind at the top of the column. The monooxygenase is eluted with 10 mM citric
acid containing Triton DN-65 (5 mg/ml), buffered at pH 6.0 with 10 mM potassium
phosphate (Figure 13). Concentration of the eluate using
an XM-50 ultrafiltration membrane yields a pale-yellow solution of thyroidal
monooxygenase. The monooxygenase is stored on ice at 4°C.
FIGURE 13
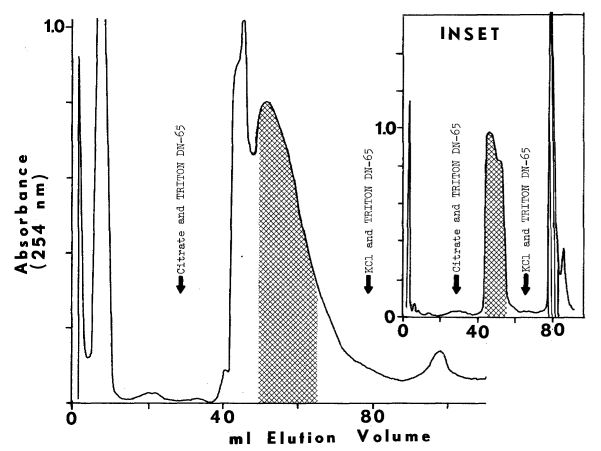
Elution of the Unidentified Microsomal Factor. A purified preparation
containing high NADPH-dependent iodinating activity (2.2 nmoles/min/mg
protein) can be obtained by chromatographing a detergent extract of acid-washed
microsomes through carboxymethyl cellulose phosphate. Chromatography of
this preparation on diethylaminoethyl cellulose acetate by the procedure
used to purify the monooxygenase causes precipitous losses in iodinating
activity. Extensive recombinant studies indicate that an essential component
remains on the diethylaminoethyl cellulose citrate column. Although a reddish
material remains at the top, and purified soluble factor binds tenaciously
to diethylaminoethyl cellulose citrate, adding soluble factor to the monooxygenase
does not restore iodinating activity. Elution of the citrate column with
0.3 M KCl containing 5 mg/ml Triton DN-65 (Figure 13)
yields a fraction that, in combination with the soluble factor and monooxygenase,
does restore iodinating activity (Table 5). This fraction
has not been further characterized. It is usually stored at 4°C without
further manipulation.
RECONSTITUTION OF IODINATING ACTIVITIES
As the preceding results imply, the NADPH-dependent
iodinating system requires at least four components. When assayed separately
the components account for only 2.6 percent of the activity present in
acid-washed microsomes. Since nearly one-third of the original activity
is reconstituted with the purified components (Table 5),
at least 12-fold synergism is possible. Iodination catalyzed by the reconstituted
system is optimized with respect to cysteamine, but concentrations of other
components are added in roughly the same proportion as they are isolated.
PROPERTIES OF PURIFIED COMPONENTS
Soluble Factor. This component is quite sensitive to changes
in pH, especially during chromatography on carboxymethyl cellulose. At
pH 6.0 or below the factor is rapidly inactivated. A sample (pH 7.2) chromatographed
at pH 7.2 contains microsomal and soluble contaminants, since cytochrome
c
and its reductase are detected both enzymically and upon electrophoresis
in gels containing sodium dodecyl sulfate and dithiothreitol. By carefully
manipulating pH (Figure 12), a fraction essentially
free of microsomal components and the smaller chromophore is obtained.
Microsomal components and possibly thyroglobulin elute in the void volume.
A chromophore electrophoretically similar to cytochrome c elutes
only at higher pH.
Purified soluble factor, comprising roughly 0.4 percent of total thyroidal protein (Table 1). contains a prosthetic group very similar to protoporphyrin IX from hemoglobulin (Figure 14). Derivatives of soluble factor and hemoglobulin absorb maximally at the following wavelengths (nm):

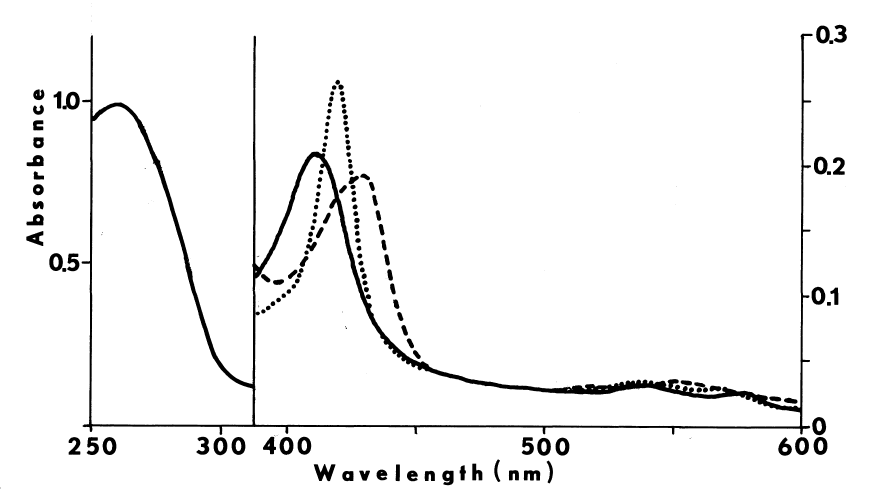
The reduced pyridine hemochromogen (Figure 15) also resembles heme B, as the following wavelengths (nm) for maximal absorbance indicate:

The oxy-pyridine derivative (Figure 15), like that of horseradish peroxidase [10], absorbs maximally at 401 nm. Based upon these similarities, the extinction coefficient for hemoglobulin at 542 nm (14.4/mM/cm) relative to that at 415 nm (131/mM/cm) [118] is used to correct estimates of protein for the absorbance of heme. Treatment of soluble factor with trichloroacetic acid yields values of protein identical to those corrected spectrally.
FIGURE 15
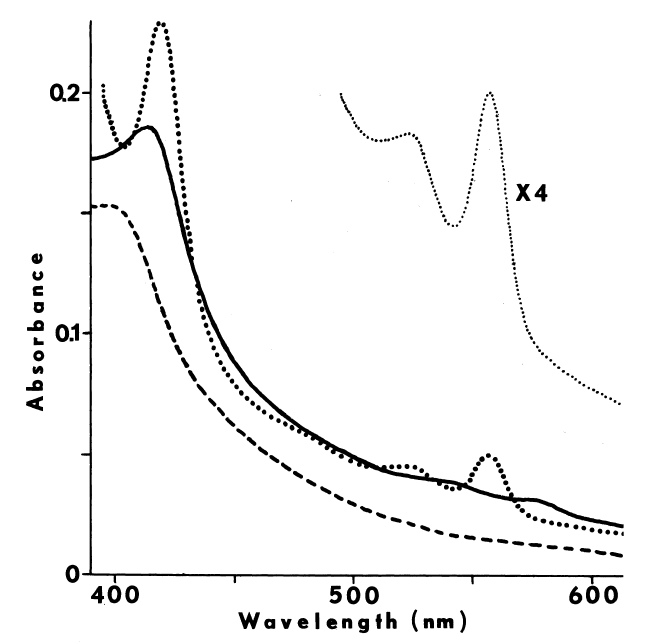
Titrating the soluble factor with half-cystinyl P-9 blue-shifts maxima in the spectra of both the oxy- and deoxy-derivatives from 413 to 409 nm and from 429 to 421 nm, respectively. Both phenomena approach saturation at 2.6 µM peptide (Figure 16).
FIGURE 16
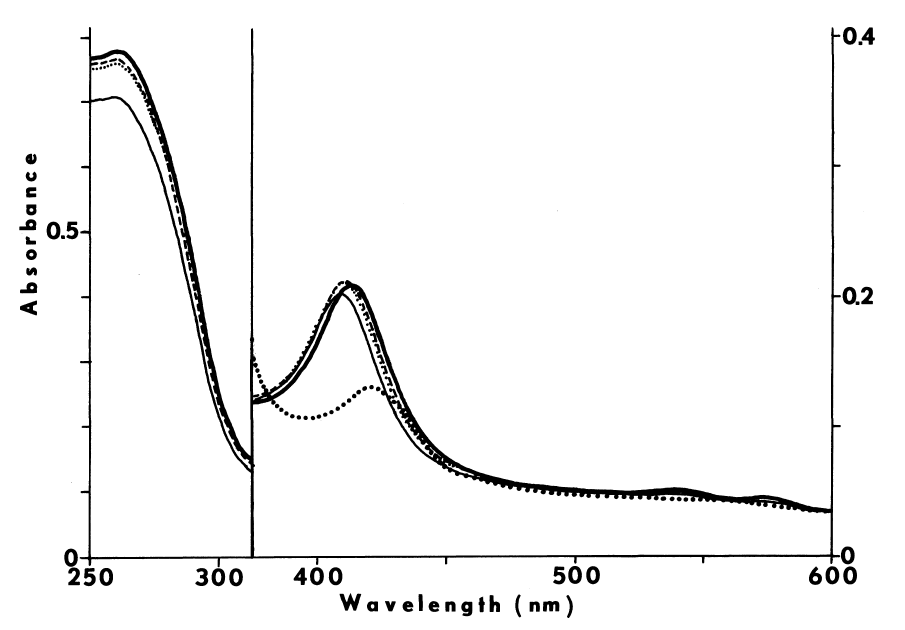
Spectral titration of soluble factor with half-cystinyl P-9. A solution containing 0.16 mg/ml of freshly thawed soluble factor (bold line) is titrated with increasing concentrations of the peptidyl substrate: 0.46 µM (broken line); 0.78 µM (lightly dotted line); and 2.6 µM (light line). In contrast to the deoxy-derivative (Figure 15), soluble factor reduced with sodium dithionite in the presence of peptidyl substrate absorbs only slightly at 429 nm. Absorbance of the deoxy-derivative at 421 nm is also quenched by the peptide (boldly dotted line).
The molecular weight of soluble factor (63,100) is estimated electrophoretically in gels containing sodium dodecyl sulfate and dithiothreitol (Figure 17). Using this value, the extinction coefficient of oxy-hemoglobulin at 415 nm (131/mM/cm), and estimates of protein corrected spectrally, the molar heme to protein ratio for highly purified soluble factor is 1.05 (Figure 12). The peak fraction is flanked by material displaying lower and higher heme to protein ratios.
FIGURE 17
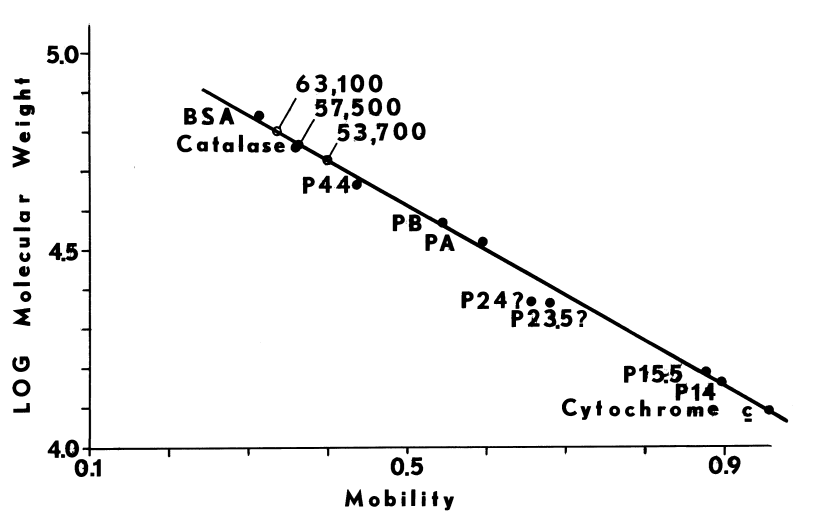
Estimation of molecular weights. Molecular weights for soluble factor and the hepatic and thyroidal monooxygenases are extimated electrophoretically on polyacrylamide slab-gels [103,117]. Bovine serum albumin, catalase, various peptic fragments of bovine serum albumin, and cytochrome c serve as standards. Soluble factor migrates as a protein of molecular weight, 63,100. Monooxygenase from the liver is estimated at 57,500 g/mole. Aged hepatic monooxygenase and the thyroidal monooxygenase both seem somewhat smaller at 53,700 g/mole.
Amino-acyl composition of the soluble factor when compared to thyroid peroxidase indicates that the former is enriched in hydrophilic residues, especially glutamate and lysine (Table 6). The soluble factor also contains considerably less proline, half-cystine, and tyrosine. Recovery of amino acids is somewhat greater than expected from corrected protein determinations: 62 µg amino acids from 53.6 µg of protein (biuret). Protein determinations are, therefore, underestimated by 13.5 percent. Assuming complete recovery of amino acids, the ratio of heme to protein in the 7 to 17 ml eluate (Figure 12) is between 0.91 and 1.01 mole of heme per mole (63,100 g) protein. The ratio of absorbances, 413 to 280 nm, is from 0.57 to 0.63.
TABLE 6
Thyroidal Monooxygenase. Thyroidal monooxygenase behaves chromatographically as the hepatic enzyme does on diethylaminoethyl cellulose (Figure 13). Absorbance of the thyroidal preparation at 254 nm is, however, greater than expected for the low activity. Also flavin is barely discernible in thyroidal preparations in contrast to the intense yellow of hepatic monooxygenase. Electrophoretically the thyroidal monooxygenase displays only one major band on gels containing both sodium dodecyl sulfate and dithiothreitol. A band migrating similarly in the hepatic preparation is probably a degradation product of the monooxygenase, since the band increases in intensity at the expense of pure enzyme upon aging at 4°C. The molecular weight of the fragment and thyroidal monooxygenase is estimated at 53,700 g/mole (Figure 17).
Unknown Microsomal Component. Very little is known about the
component of the NADPH-dependent system eluted from diethylaminoethyl cellulose
with 0.3 M KCl. Although present, protein in this fraction disperses electrophoretically
over a wide range of microsomal proteins. In addition, cytochrome c
reductase activity is detectable in this fraction. The chemical nature
of this factor and its behavior in the NADPH-dependent iodinating system
remains largely unexplored.
ACTIVITIES OF INDIVIDUAL COMPONENTS
Soluble Factor. Alone, soluble factor displays relatively little
NADPH-dependent iodinating activity (Tables 1,5).
Chromatography on carboxymethyl cellulose disrupts peroxidation of iodide
catalyzed by thyroid peroxidase as well (Figure 12).
Both activities are reconstituted (Table 5) through the
combined actions of soluble factor and the microsomal components. The latter
components may be somewhat more labile to peroxide than soluble factor,
since preincubation of soluble factor with peroxide for 1 minute before
addition of microsomal components produced faster rates of periodide generation
than if the order of addition is reversed.
Thyroidal Monooxygenase. Like the hepatic enzyme, thyroidal monooxygenase is activated by primary alkylamines and alkylguanidines, as indicate below:

Activity in microsomes from porcine thyroid is seldom observed in the absence of activator. N-oxygenation of dimethylaniline and methimazole-dependent oxidation of DTNB are readily demonstrable when n-octylamine or n-dodecylguanidine is in the the assaying medium. Upon purification, activity is discernible in both activated and untreated preparations.
Acceptable losses of the hepatic or thyroidal monooxygenase occur during purification on diethylaminoethyl cellulose (Figure 13). The following specific activities of the hepatic enzyme indicate 88 percent purity and 100-fold purification:

No other flavoproteins contaminate the hepatic
monooxygenase, for the activity to flavin ratio is that of pure enzyme.
This may also be true of the thyroidal preparation, since there is no detectable
cytochrome c reductase activity. Both monooxygenase preparations
are contaminated with catalase; the thyroidal preparation contains a 140-fold
catalytic excess of catalase.
REFINEMENTS TO THE ASSAY OF NADPH-DEPENDENT IODINATION
Although useful in elucidating the components of the NADPH-dependent iodinating system, the standard assay is not without problems.
Inhibition by Half-Cystinyl P-9. Half-cystinyl P-9 inhibits NADPH-dependent iodination of thyroidal proteins, at the concentrations employed in the standard assay (Figure 18). Unlike microsomes, the reconstituted system probably contains very little endogenous substrate. Apparent Km for half-cystinyl P-9 using the reconstituted system is 1.6 µM (Figure 19).
FIGURE 18
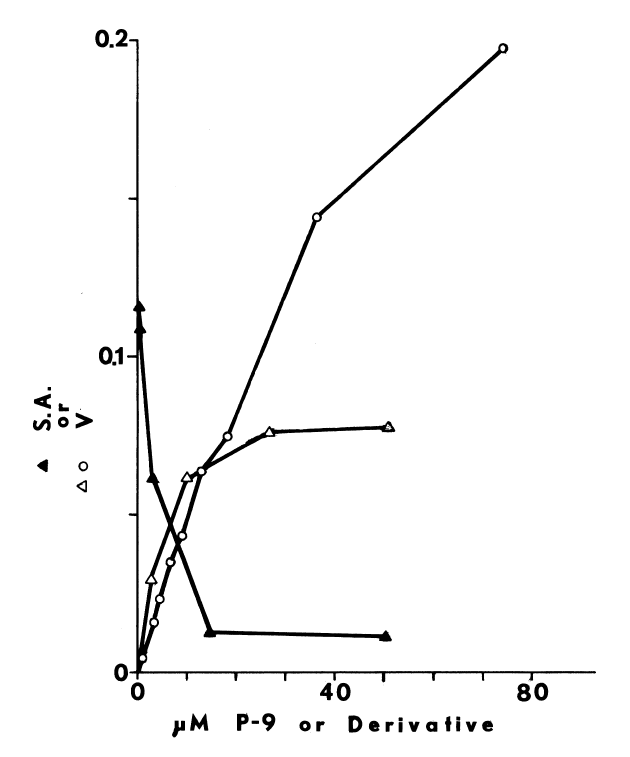
Iodination of lysylglutaryl P-9. Glutamyl-tyrosyl-glutamate is not substrate for the NADPH-dependent system (Figure 4), but the N-acetylated derivative is iodinated (Figure 5). A similar sequence in the P-9 peptide contains a free amino group:
lysylglutamyltyrosylglutamyl
Blocking the free amino groups of P-9 with lysylglutamyl groups profoundly alters behavior of this peptide in the NADPH-dependent system. The modified peptide is iodinated at a much faster rate (Figure 18), and displays a higher apparent Km of 90.9 µM (Figure 19).
FIGURE 19
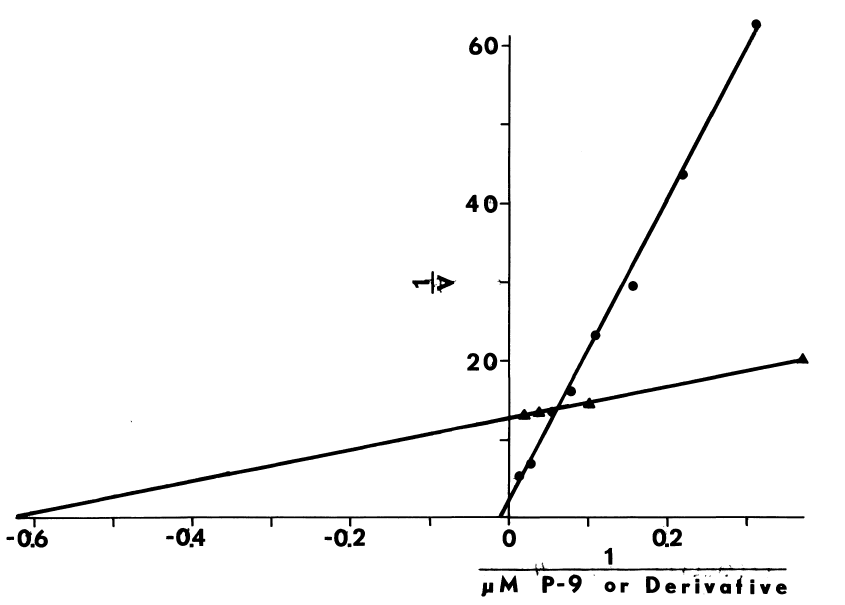
Double reciprocal plots of NADPH-dependent iodination catalyzed by the reconstituted system. Iodination is assayed as described in the legend of Figure 18. Velocities (V) are expressed in nmole/minute/ml of the reconstituted system. Iodination of lysylglutaryl P-9 or half-cystinyl P-9 are represented by circles and triangles, respectively.
Lysylglutaryl P-9 also has technical advantages over half-cystinyl P-9. Unlike half-cystinyl P-9, it remains soluble after the mixed disulfides are reduced. As a consequence, assays performed in the presence of glutathione need not be contaminated with cysteine reductively liberated from the peptide. Increasing the hydrophilicity of the peptide also improves precipitation with acetone. Enhanced recovery of product results in more accurate determinations of iodinating activity.
Other Aminoethanethiols. Other endogenous aminoethanethiols, with one notable exception, are no more effective than cysteamine in stimulating NADPH-dependent iodination. Only a digest of dephosphocoenzyme A (MATERIALS) has a more pronounced effect (Table 7). Of the possible hydrolytic products, AMP stimulates only at low concentrations of iodide; pantetheine is only slightly more effective than cysteamine.
TABLE 7
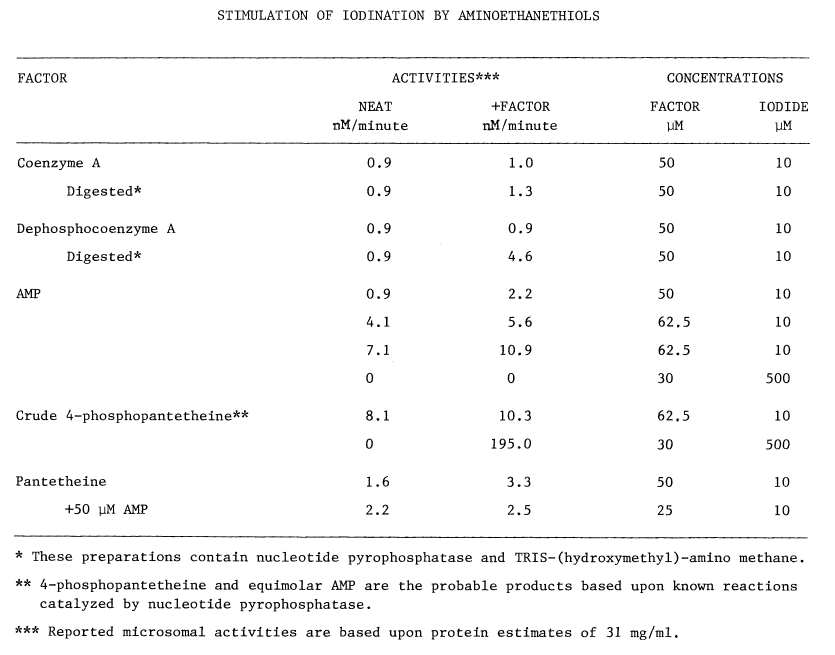
OXIDATION OF IODIDE CATALYZED BY THE HEPATIC MONOOXYGENASE
Iodide Kinetics. Oxidation of iodide catalyzed by the hepatic monooxygenase is influenced by many xenobiotic and endogenous factors. Crude enzyme is half-saturated by 1 mM iodide when assayed in 0.1 M potassium phosphate buffer (Figure 20). At this concentration of buffer, maximum velocity for the oxidation of iodide is indistinguishable from that for methimazole. At lower concentrations of phosphate, however, rates are somewhat slower. Perchlorate, an agent that blocks the accumulation of iodide in the thyroid, inhibits instead of activates monooxygenase-catalyzed oxidation of iodide.
FIGURE 20
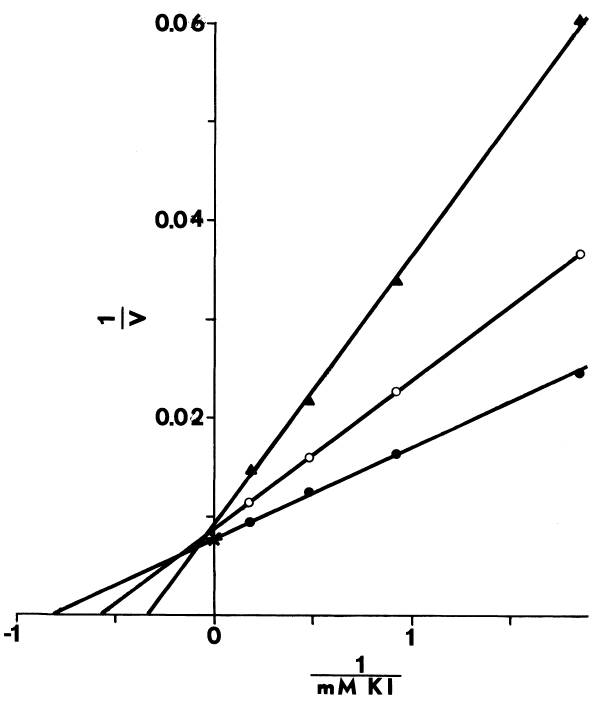
Glutathione in the crude system also stimulates the oxidation of iodide (Figure 21). Extensive chromatography of the hepatic monooxygenase on diethylaminoethyl cellulose yields enzyme that is half-saturated with 0.57 mM iodide and is inhibited rather than activated by glutathione (Figure 22). Binding of glutathione and iodide with the monooxygenase seems mutually exclusive in the purified system, while glutathione in the crude system appears to remove a competitive inhibitor (Figures 21,22).
FIGURE 21
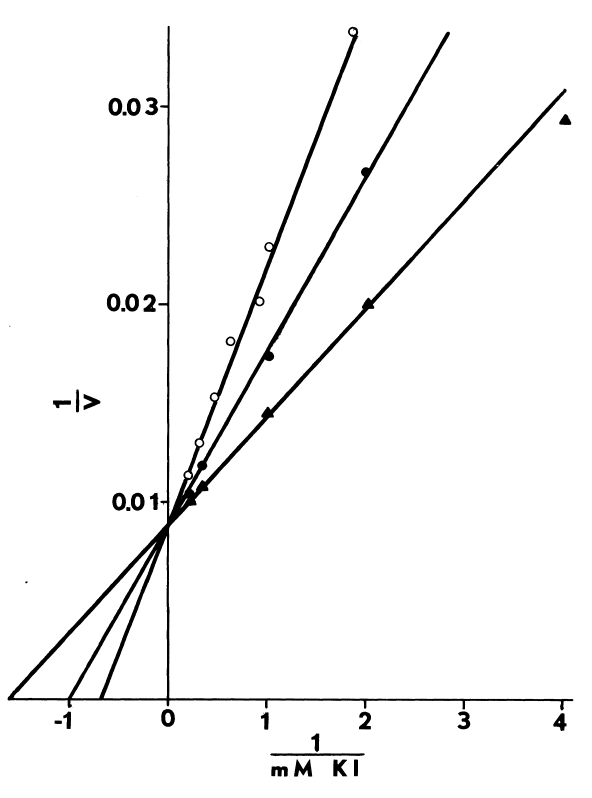
Effect of glutathione and calcium ion on iodide-dependent uptake of oxygen catalyzed by the crude hepatic monooxygenase. Uptake of oxygen is measured polarographically as previously described [78]. In addition to the glucose-6-phosphate generating system and 50 mM potassium phosphate buffer (open circles), media contain 0.25 mM glutathione alone (filled circles), or 0.25 mM glutathione and 10 µM calcium acetate (triangle). Velocities (V) in these double reciprocal plots are in nmole/minute/mg protein.
FIGURE 22
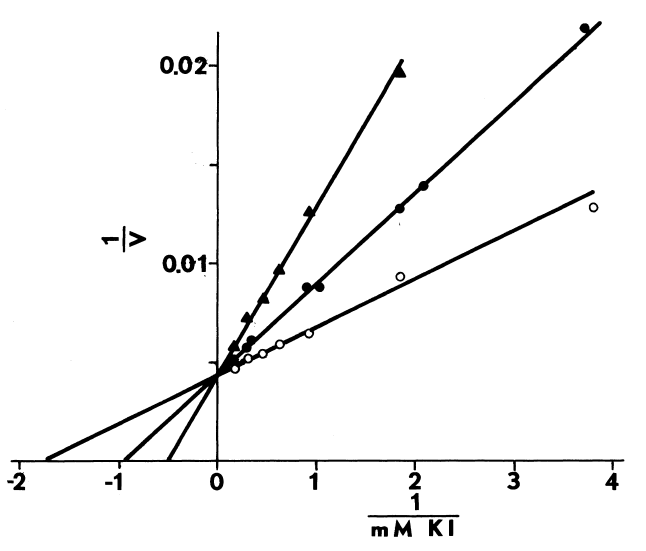
Effect of glutathione on iodide-dependent uptake of oxygen catalyzed
by the highly purified hepatic monooxygenase. Uptake of oxygen is determined
as previously described [78]. Open circles represent
activities in the absence of glutathione. Activities in the presence of
0.25 or 2.5 mM glutathione are depicted by filled circles and triangles,
respectively. Velocities (V) in these double reciprocal plots are in nmole/minute/mg
protein.
Calcium and Octylamine Effects. Low concentrations of calcium, 10 µM, potentiate the stimulatory effect of glutathione in the crude system (Figure 21). Response to calcium is altered as the monooxygenase is purified on diethylaminoethyl cellulose. The same concentration of calcium when added to media lacking glutathione is inhibitory (Figure 23).
FIGURE 23
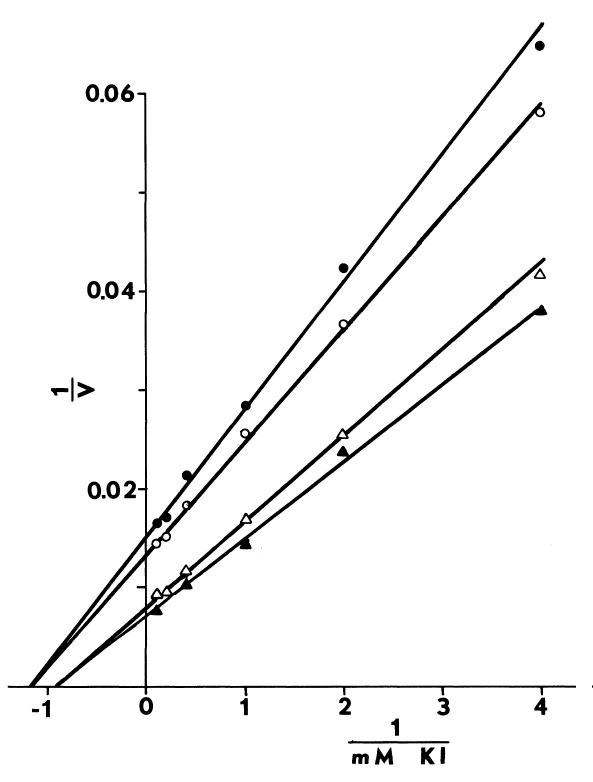
Effect of calcium ion, octylamine, and cysteamine on iodide-dependent uptake of oxygen catalyzed by crude hepatic monooxygenase. Uptake of oxygen is measured polarographically as previously described [78]. Velocities (V) in these double reciprocal plots are expressed in nmole/minute/mg protein. Ten µM calcium acetate (filled circle), 1.25 mM octylamine sulfate (open triangle), or 1.25 mM octylamine sulfate and 10 µM cysteamine (filled triangle) have the indicated effects on unsupplemented activities (open circle). Glutathione is not added to these media.
As with other monooxygenase-catalyzed reactions, octylamine nearly doubles the iodide-dependent uptake of oxygen. Cysteamine, 10 µM, in the presence of octylamine is slightly stimulatory (Figure 23).
Cysteamine Kinetics. Cysteamine, being the only known endogenous
substrate for hepatic monooxygenase, is a possible acceptor in the formation
of sulfenyl iodide. Apparent Km obtained with recrystallized
cysteamine, 38 µM, is somewhat lower than previously reported [81].
As before, interaction of only one mole of cysteamine with the enzyme is
rate limiting.
Protection of NADPH by Cysteamine. Cysteamine prevents the irreversible
oxidation of NADPH dependent upon iodide and the hepatic monooxygenase.
The protective effect is saturable in cysteamine, and does not exceed 25
percent of the unprotected rate at 0.5 mM iodide. The amount of cysteamine
providing half-maximal protection is 18 µM (Figure
24).
FIGURE 24
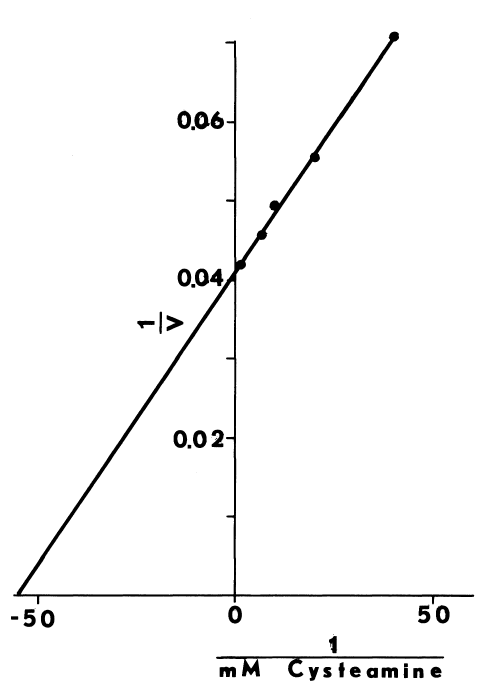
Protection of NADPH by cysteamine in reactions catalyzed by the hepatic
monooxygenase. Iodide and monooxygenase-dependent destruction of NADPH
in the presence of the glucose-6-phosphate generating system [78]
is monitored at 340 nm as described in Table 8. At 0.5 mM potassium iodide
and in the abscence of glutathione, cysteamine prevents loss of absorbance
at 340 nm. Velocities (V) in these double reciprocal plots indicate the
percentage of the rate of destruction prevented by a variety of cysteamine
concentrations.
Effects of Aminoethanethiol and Iodide on the Monooxygenase. Hepatic FADcontaining monooxygenase is rapidly inactivated by nucleotide pyrophosphatase (Figure 25). The pyrophosphatase when incubated with dephosphocoenzyme A, however, produces a substance that together with 0.5 mM iodide prevents inactivation of the monooxygenase. Dephosphocoenzyme A, AMP, and pantetheine separately or in combination with 0.5 mM iodide do not protect the monooxygenase from inactivation by the pyrophosphatase. Under these conditions iodide and the hydrolysate in the abscence of methimazole do not appreciably affect uptake of oxygen.
FIGURE 25
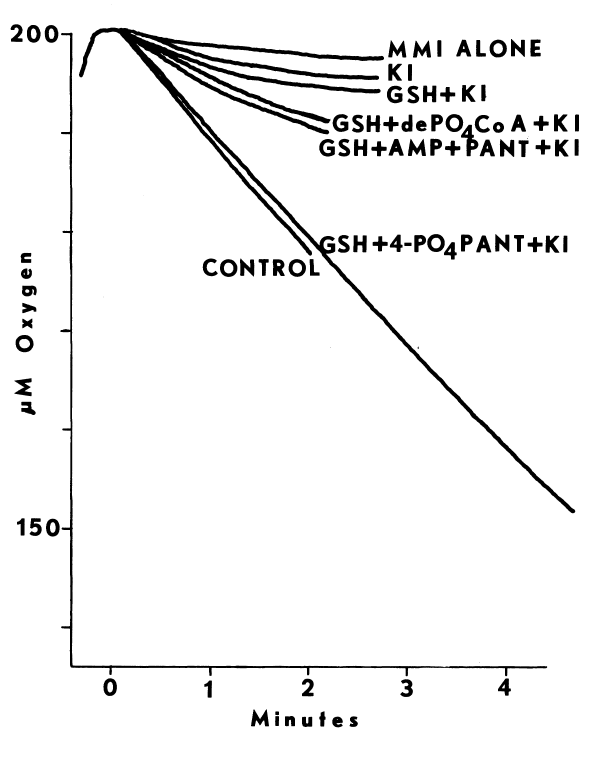
Destruction of methimazole and monooxygenase-dependent uptake of
oxygen by nucleotide pyrophosphatase. Uptake of oxygen catalyzed by
the highly purified hepatic monooxygenase is determined polarographically
as previously described [78]. Sufficient nucleotide pyrophosphatase
to inactivate the monooxygenase in three minutes is added to all media
except the control. All media contain mmolar magnesium chloride. The following
additions from top to bottom fail to prevent inactivation of methimazole-dependent
uptake of oxygen: none (MMI ALONE); 0.5 mM potassium iodide (KI); mmolar
glutathione and 0.5 mM potassium iodide (GSH + KI); mmolar glutathione,
50 µM dephosphocoenzyme A, and 0.5 mM potassium iodide (GSH + dePO4CoA
+ KI); or mmolar glutathione, 50 µM AMP, 50 µM pantethine,
and 0.5 mM potassium iodide (GSH + AMP + PANT + KI). An enzymic hydrolysate
of dephosphocoenzyme A, approximately 50 µM, in combination with
0.5 mM potassium iodide and mmolar glutathione (GSH + 4-PO4PANT + KI) provides
complete protection.
Recombinant Studies with Purified Peroxidase. The effects of various endogenous factors and enzymes on iodide-dependent oxidation of NADPH are presented in Table 8. As before, cysteamine (CSH) prevents the irreversible oxidation of NADPH catalyzed by the hepatic monooxygenase (MO). Thyroid peroxidase (TPO) reverses this protection, but only in the absence of tyrosine (TYR). Glutathione (GSH) protects NADPH completely. Protection afforded by glutathione and cysteamine is gradually lost. In the presence of purified thyroid peroxidase, NADPH protected by cysteamine is initially more labile, but after 4 to 5 minutes is less labile than when peroxidase is lacking.
TABLE 8
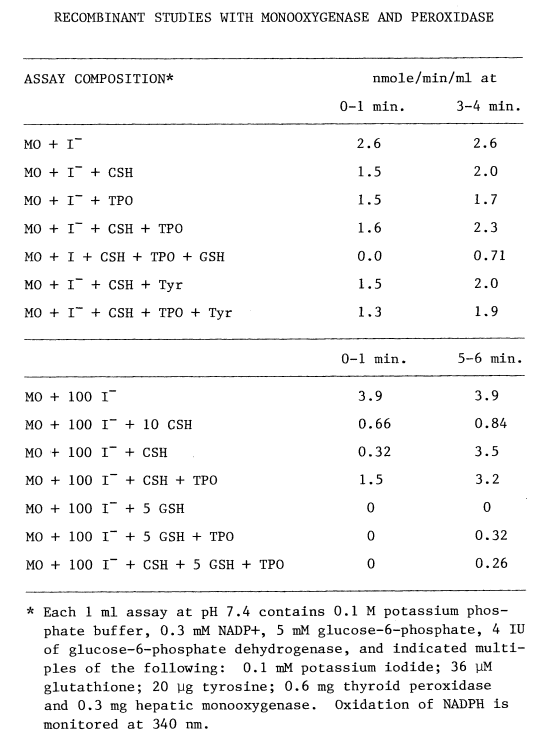
Diminished protection at early times and enhanced protection later are both saturable with peroxidase (Figure 26).
FIGURE 26
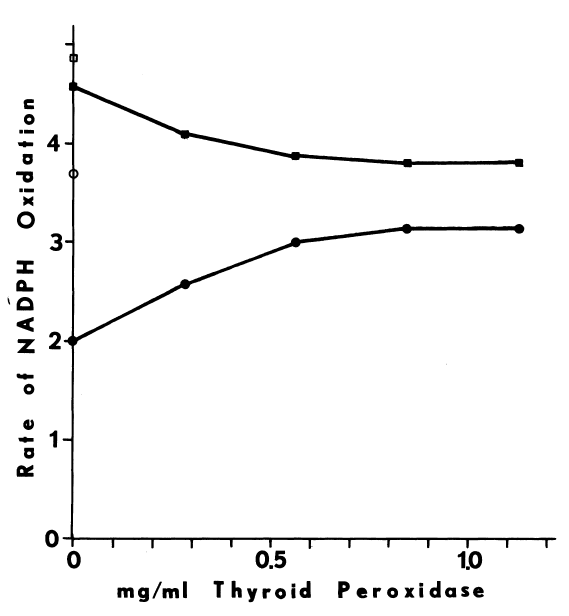
Destabilization of intermediary iodines with thyroid peroxidase. The system contains 0.6 mg/ml of hepatic monooxygenase and 100 µM iodide. Destruction of NADPH is monitored as described in Table 8. Open symbols depict rates without cysteamine. Filled symbols represent rates of NADPH destruction (nmole/minute) in media containing 36 µM cysteamine and varying concentrations of thyroid peroxidase. Circles or squares reflect initial rates, up to one minute, and rates of destruction after four minutes, respectively.
DISCUSSION
In reviewing what is currently known about the biosynthesis of thyroxine, it is obvious that the peroxide-dependent model does not satisfactorily depict the in vivo iodinating system. Indeed, the source of peroxide purportedly endogenous to the thyroid remains unidentified. NADPH-cytochrome c reductase is the most likely source of peroxide heretofore explored [7]. However, antibodies to this enzyme suppress iodination by only 50 percent, and do not affect NADH-dependent activity. When supplemented with both reduced pyridine nucleotides, iodination catalyzed by the particulate fraction is inhibited less than 22 percent by antibodies to the reductase [122]. Another mechanism for iodination in the thyroid is clearly indicated. Results presented in this dissertation comprise the first definition of a system that is, at once, dependent upon reduced pyridine nucleotides and independent of peroxide. Unlike the peroxide-dependent system, the one proposed herein retains most, if not all, characteristics of the crude system as it is resolved, purified, and reconstituted.
Obtaining active tissue is a major obstacle in studies of NADPH-dependent iodination. Since chilling immediately after exsanguination of the hog protects thyroidal activities, NADPH-dependent iodination is probaby heat-labile. The thyroid, by storing thermogenic hormone, may be especially prone to a post-mortem rise in temperature. Loss of NADPH-dependent iodinating activity does parallel the inactivation of thyroidal and hepatic monooxygenases, and implicates the monooxygenase in the iodinating pathway.
Conditions employed in monitoring this iodinating activity differ markedly from those previously used. Glutathione and cysteamine are included to fully stimulate NADPH-dependent iodination and eliminate iodination dependent upon peroxide (Table 4, Figures 8,9). Glutathione apparently alters availability of peroxide or inhibits peroxidase-catalyzed iodination directly. Concentrations of these two thiols optimal in the media differ from those measured in the thyroid (Table 2). Compartmentalization of thiols within the thyroid may account for these differences, for follicular epithelium comprises the following fraction of the total thyroidal volume [28]:

Assuming most of the thiol is present within the follicular cell, NADPH-dependent iodination is optimal at endogenous concentrations of cysteamine and glutathione.
Other evidence substantiates this distribution of thiols. The thiol to disulfide ratio in protein from whole thyroids is 0.11. This is considerably higher than the ratio reported for thyroglobulin, 0.05 [73]. Although a more reductive environment seems likely in the follicular cell, spurious oxidations of thiols during purification of thyroglobulin could create a lower apparent ratio.
Thyroglobulin in the homogenate may also detract from the significance of cytosolic enzymes. Specific activities of glutathione reductase, glutathione peroxidase, catalase, and thioltransferase (Table 2) average 20 times lower in the porcine thyroid than in rat liver [107,106,109,123]. Considering the gross contamination of the cytosolic compartment with protein from the lumen, the follicular cell contains a near normal complement of enzymes. As in most other plant and animal tissues [73], a reductive intracellular environment is probably maintained.
In addition to being less sensitive to glutathione, the NADPH-dependent system is much more selective for proteinaceous substrates than iodination dependent upon peroxide. Relative sensitivities to glutathione and specificities for peptides are illustrated in Figure 4. Unlike the peroxide-dependent system [7,15,16,48,49,54,59], those systems dependent upon NADPH do not iodinate peptides containing vicinal amino groups readily; glutamyl-tyrosyl-glutamate (Figure 4) and half-cystinyl P-9, containing similar sequences, are inhibitory (Figure 18). The N-acetylated derivative of the tripeptide is readily iodinated (Figure 5). Furthermore, upon blocking the lysyl residues in half-cystinyl P-9 with lysylglutaryl Schiff bases and reducing with borohydride, the maximal rate of iodination in the reconstituted system increases (Figures 18,19). Figure 5 also indicates that the free amino group is not solely responsible for the inhibition, since even the N-acetylated tripeptide becomes inhibitory as microsomes age at 4°C. Aging apparently removes a factor that influences the specificity of NADPH-dependent iodination for peptidyl substrates.
As might be expected from structural considerations, much of the inhibition by peptides is attributable to ionic interactions with the system. Potassium chloride (0.1 M) restores some activity lost upon aging (Figure 5), but most of the inhibition caused by the modified tripeptide is unaffected. Calcium ion (10 µM) in combination with 10 mM magnesium acetate eliminates the remaining inhibition. Presumably, chloride and calcium ion influence association of the modified peptide with the iodinating system. Iodination may change the pKa of the tyrosyl residue [23], and create a more effective chelator for divalent cations. While chelation of product could account for the observed stimulation, such an effect is highly dependent upon the nature of the substrate, and may be of little physiological significance.
Ionic interactions of a more complex nature are suggested in studies with an unmodified peptide (Figure 9). Like inhibition by the modified tripeptide, aged microsomes are less active than fresh when half-cystinyl P-9 is the substrate. Glutathione at high concentrations, 7 to 15 mM, enhances iodination catalyzed by aged microsomes disproportionately. Based upon structural considerations (Figure 27) glutathione could, conceivably, influence association of the peptide with aged microsomes.
FIGURE 27
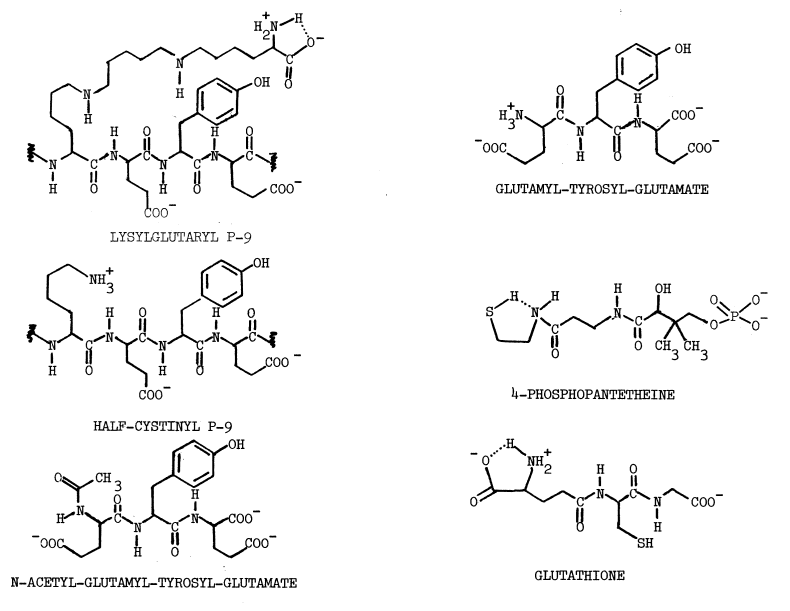
Structures of substrates and effectors involved in NADPH-dependent iodination.
Direct modification of certain tyrosyl peptides also augments iodinating activities (Figures 18,19). With the reconstituted system lysylglutaryl P-9 displays faster rates of iodination than other peptides tested, but microsomal activities (Table 7) are lower than those obtained with half-cystinyl P-9 (Tables 1,5). This suggests that the sequence analogous to N-acetyl-glutamyl-tyrosyl-glutamate is sterically hindered in the modified P-9, allowing only those sequences with lower affinity and higher turnover to interact. The following sequence within P-9 is quite similar to the one most hormonogenic in thyroglobulin [24,120]:
C-Histidyl-arginyl-arginyl-seryl-tyrosyl-glutamyl-tyrosyl-leucyl-N
P-9
-alanyl-seryl-tyrosyl-glutamyl-aspartyl-C
Thyroglobulin
Compared to the endogenous substrate this sequence is inverted, N-terminus for C-terminus. This difference may explain the apparent inability of the modified peptide to compete with microsomal iodine acceptors.
Although NADPH-dependent iodination catalyzed by microsomes is predictably selective for tyrosyl substrates, that catalyzed by the post-microsomal supernatant is not. When microsomes are removed at alkaline pH, this fraction preferentially catalyzes iodination of glutamyl-tyrosyl-glutamate (Figure 4). Half-cystinyl P-9, in contrast, is readily iodinated in reactions catalyzed by the acidic supernatant. An inhibitor of NADPH-dependent iodination is removed with microsomes under acidic conditions, for the supernatant is much more active with half-cystinyl P-9 than the original homogenate (Table 1). Under more alkaline conditions the substance apparently remains in the supernatant (Figure 4). Ironically, the alkaline supernatant catalyzes iodination of glutamyl-tyrosyl-glutamate more effectively than microsomes, so the substance may be more regulatory than inhibitory. Subcellular redistribution of other components with changes in pH complicates interpretation of these results. Since the specific activity of the acidic supernatant is considerably lower than microsomes (Table 1), these anomalies in the soluble fraction are not studied further.
Alkaline pH removes a hemoprotein from microsomes that have been extensively washed under acidic conditions (Table 1). Removal of this soluble factor diminishes activity remaining in frozen microsomes (Table 5). Purification on carboxymethyl cellulose yields a factor which is at least 90 percent pure, based upon a molar heme to protein ratio of one and a molecular weight of 63,100 (Figure 17). Having no appreciable activity of its own (Tables 1,5), soluble factor is essential for both NADPH-dependent iodination (Table 5) and peroxide-dependent oxidation of iodide. Lability of this factor at low pH on carboxymethyl cellulose is consistent with the previously reported dissociation of thyroid peroxidase into apoenzyme and prosthetic group [124]. Procedures presented herein differ in that the pH is carefully controlled at 7.0. Yields of this highly purified hemoprotein (Table 1), are at least 1,000 times greater than those reported for thyroid peroxidase [10].
Identity of the soluble factor with a subunit of thyroid peroxidase is inferred. Physical and chemical evidence, as well as the aforementioned enzymic evidence, support this contention. Since the soluble factor has a molecular weight only slightly larger than the 60,000 subunit of the peroxidase [10], the amino acyl compositions of these two proteins are compared (Table 6). The 24,000 subunitpresent in the peroxidase [10], but not in soluble factorprobably accounts for most differences. Soluble factor, nonetheless, seems particularly enriched in lysyl and glutamyl residues. The slightly higher molecular weight and greater solubility probably results from the mild purification conditions used. Unlike thyroid peroxidase [10], soluble factor is removed from the membrane by nonproteolytic methods. Conceivably, proteolysis produces a fragment of soluble factor lacking hydrophilic residues and appearing integral to the membrane.
The spectra of soluble factor and its various derivatives (Figures 14,15) are virtually indistinguishable from those of hemoglobulin and horseradish peroxidase [10,118,121]. This establishes protoporphyrin IX as the prosthetic group in soluble factor. Although small spectral differences in pyridine hemochromogens are evident [10], thyroid peroxidase also contains protoporphyrin IX [7]. Tyrosyl substrates contaminating even the most highly purified thyroid peroxidase [10], probably cause these deviations, since half-cystinyl P-9 shifts the spectrum of soluble factor (Figure 16) to that of thyroid peroxidase [7,65]. Tyrosyl substrate may be the major contaminant in soluble factor as well, since maximal absorbance is at a lower wavelength, 413 nm, than reported for oxy-hemoglobulin, 415 nm. Concentrations of half-cystinyl P-9 causing the spectral transitions to 409 nm (Figure 16), are roughly those expected from kinetic measurements (Figure 19). This implicates soluble factor as the iodotransferring catalyst in the NADPH-dependent pathway.
Other parameters of soluble factor attest to its purity when compared with thyroid peroxidase. Absorbance of a 1 mg/ml solution of soluble factor at 411 nm is 2.1 (Figure 12). This is nearly 27 times that reported by others for peroxidase solubilized by nonproteolytic methods [65]. The ratio of absorbances at two wavelengths, 413 to 280 nm, is also higher: 0.63 compared to 0.54 [10]. This reflects the higher molar heme to protein ratio of 1.0. Soluble factor, therefore, appears to be a subunit of thyroid peroxidase that is at once intact, highly purified, and iodotransferring.
The only other characterized component of the NADPH-dependent iodinating system is the thyroidal monooxygenase. In crude preparations, NADPH-dependent iodination has many features in common with reactions catalyzed by the hepatic microsomal FAD-containing monooxygenase: activity sustained by NADH is 80 percent of that obtained with NADPH [13,89,125], NADP+ is inhibitory [12,78], bound flavin is required [13,126], and activities are labile to heat [9,11,15,17,59,61,81,100] and sonication [15]. It is not surprising, then, that a similar enzyme is found to be an essential component of the thyroidal iodinating system (Table 5).
The thyroidal monooxygenase is catalytically and chromatographically (Figure 13) identical to the hepatic enzyme. Chromatographic behavior may have bearing on previous studies of thyroid peroxidase [10,16,64], for monooxygenase elutes from diethylaminoethyl cellulose under roughly the same conditions as thyroid peroxidase.
Thyroidal monooxygenase also resembles the hepatic enzyme electrophoretically. In gels containing sodium dodecyl sulfate and dithiothreitol, the purified thyroidal enzyme migrates as a single major band having a molecular weight of approximately 53,700 (Figure 17). While freshly purified hepatic monooxygenase migrates as a protein of higher molecular weight (57,500), a band at the lower molecular weight is observed in preparations that have been stored for several days at 4°C. Activities are given priority in these initial studies, so the electrophoretic results reported are obtained with aged preparations of thyroidal monooxygenase. Presumably, fresh preparations contain thyroidal monooxygenase of higher molecular weight. The significance of this proposed degradation in other aging phenomena (Figures 5,9) has not been determined.
As noted before, propylthiouracil and methimazole, as well as other thioureas, thiouracils, and mercaptoimidazoles are substrates for the hepatic monooxygenase [78,79,81,88,89]. The antithyroidal potency of these compounds is well-documented [127]. Table 3 indicates that other substrates for the hepatic enzyme [81,126,128] are also inhibitors of NADPH-dependent iodination. Inhibition is roughly that expected based upon the Km for the hepatic monooxygenase and the degree of saturation in cysteamine at 10 µM iodide. Dimethyloctylamine, having a higher Km [126] is less inhibitory (Table 3). Dimethylaniline, pentamethylene sulfide, and o-dithiane all have Kms of less than 3 µm, but the disulfide only inhibits iodination by 50 percent. The obviating aminoethanethiol could be saturating in studies using the disulfide, since cystamine generates cysteamine in reactions with proteinaceous sulfhydryls. Alone, higher concentrations of cysteamine inhibit NADPH-dependent iodination. Clearly, substrates for the monooxygenase, with the possible exception of low concentrations of cysteamine (Figure 8), should be excluded when assaying NADPH-dependent iodinating activity.
The obviating effect of cysteamine and cystamine is also observed, in vivo. Upon exposure to either compound, thyroids previously blocked by thiouracil accumulate more iodide. When added alone 1.4 µM methimazole or 71.7 µM cysteamine half-inhibit iodination catalyzed by slices of beef thyroid [32]. These values correlate with Kms for the hepatic monooxygenase of 5.2 µM and from 38 to 120 µM, respectively [81], and confirm involvement of the monooxygenase in thyroidal iodinations.
Although inhibition is apparent initially, cystamine increases linearity of NADPH-dependent iodination with time (Figure 6). Failure to establish absolute requirements for cysteamine in the highly purified system, though, indicates involvement of another factor (Table 5). Cysteamine could prevent metabolism of precursory aminoethanethiols. Based upon the specificity of the nucleotide pyrophosphatase and the inability of other hydrolytic products to mimic the effect, 4-phosphopantetheine is the only intermediate in cysteamine's biosynthetic pathway [106] producing greater stimulation of NADPH-dependent iodination (Table 7). Since difficulties with lysylglutaryl P-9 are ameliorated by supplementing with 4-phosphopantetheine, this compound may be a limiting component in iodinations catalyzed by thyroidal microsomes.
Metabolism of microsomal 4-phosphopantetheine to cysteamine (Figure 8) might sensitize NADPH-dependent iodination to inhibition by glutathione (Figure 9, Table 8) and N-acetyl-glutamyl-tyrosyl-glutamate (Figure 5). Phosphopantetheine (Table 7) does significantly augment iodinating activities with lysylglutaryl P-9 (Figure 27). Although glutathione under certain conditions also stimulates NADPH-dependent iodination, the multiplicity of interaction illustrated in Figure 9 prevents delineation of these effects. Elimination of 4-phosphopantetheine as a variable in thyroidal preparations may make activities of the iodinating system with these peptides more predictable.
Stimulation of iodination by aminoethanethiols suggests that the intermediate is a sulfenyl iodide. Since polarization of the bond in most sulfenyl halides favors formation of the sulfenium ion, reactions with amines usually produce the following sulfenamides instead of iodoamides [77]:
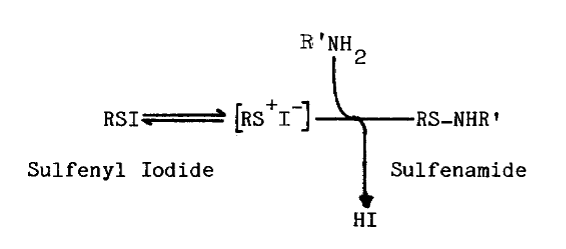
The converse, as follows, is probably true of sulfenyl iodides formed from aminoethanethiols:
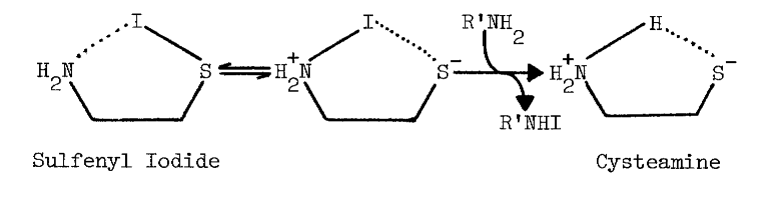
The following regeneration of precursory aminoethanethiol is a theoretically unique aspect of iodinations with these sulfenyl iodides:
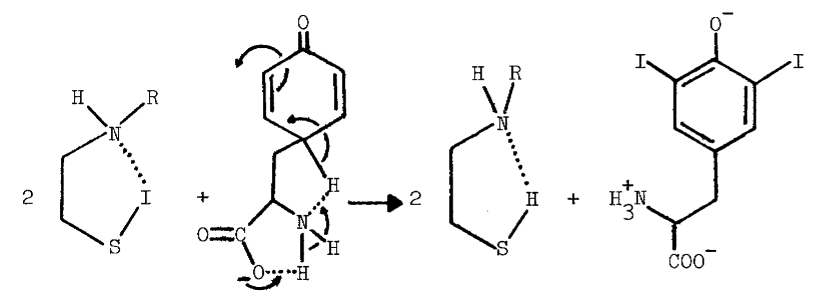
Evidence for the regeneration of cysteamine is found in reactions catalyzed by the hepatic monooxygenase and thyroid peroxidase (Table 8, Figure 26). Catalysis of iodinating reactions by this particular aminoethanethiol is of questionable physiological significance, for glutathione prevents cysteamine- and iodide-dependent destruction of NADPH catalyzed by the monooxygenase and thyroid peroxidase (Table 8). Octylamine may also regenerate substrate, since 10 µM cysteamine stimulates monooxygenase-catalyzed uptake of oxygen in the presence of the amine and iodide (Figure 23), but not when amines are lacking.
Interaction of glutathione in NADPH-dependent iodination is also far from simple. Apart from relieving inhibition by the cystinyl P-9 peptide (Figure 9), glutathione may also influence the oxidation of iodide catalyzed by the monooxygenase. Prior to purification of the hepatic enzyme on diethylaminoethyl cellulose, glutathione seems to remove a competitive inhibitor (Figure 21), but is, itself, inhitory in reactions catalyzed by the highly purified monooxygenase (Figure 22). Glutathione also stimulates iodinating activities of freshly prepared thyroidal microsomes, but becomes increasingly inhibitory as microsomes age (Figure 9). Inhibitory responses to glutathione may, therefore, indicate modifications in the NADPH-dependent iodinating system.
Calcium also favors oxidation of iodide catalyzed by the crude hepatic monooxygenase (Figure 21), but only when near physiological concentrations of glutathione are present (Figure 23). The [S]½ for iodide with highly purified hepatic monooxygenase, 0.57 mM (Figure 22), is roughly that obtained with crude monooxygenase in the presence of 0.25 mM glutathione and 10 µM calcium acetate (Figure 21). Although calcium and glutathione apparently miminize iodide requirements, high concentrations of calcium should be avoided to prevent substrate-independent uptake of oxygen (Figure 2).
Increases in substrate-independent uptake of oxygen are also observed as the monooxygenase is purified on diethylaminoethyl cellulose. Concommitantly, octylamine becomes less effective as an activator (Figure 1). Since loss of iodinating activity also occurs as the thyroidal monooxygenase is purified on diethylaminoethyl cellulose (Table 5), caution should be exercised when exposing the system to xenobiotic cations.
Alkylamines and alkylguanidines stimulate monooxygenase catalyzed reactions, but must be excluded when studying iodination. This limitation also applies to buffers containing free amines, for no activity is detected in media buffered with Tris-(hydroxymethyl)- aminomethane. Even the arginyl residues of lysylglutaryl P-9 are suspect, since another guanidine is shown to uncouple specificity without influencing the extent of iodination. The iodinated products in the presence of amines or guanidines are sensitive to bisulfite (Figure 6), a reaction characteristic of nitrogenous iodines [115].
Two compounds known to affect thyroidal accumulations of iodide must also be avoided: perchlorate and thiocyanate. These agents block uptake of iodide by the thyroid (Appendix). Perchlorate also prevents iodide-dependent destruction of NADH and NADPH [53]. Neither perchlorate nor thiocyanate is substrate for the hepatic monooxygenase. At millimolar concentrations, however, both inhibit uptake of oxygen in monooxygenase-catalyzed reactions (Figure 20). Thiocyanate also inhibits NADPH-dependent iodination catalyzed by thyroidal microsomes (Figure 10). Unlike the effect on the uptake of oxygen, inhibition of iodination is nearly complete at one mM thiocyanate (Figure 11).
Evidence presented in this dissertation is consistent with a peroxide-independent pathway for the synthesis of thyroxine. The system as resolved and purified constitutes the basal iodinating activity in the thyroid. Components apparently catalytic in the thyroidal system are a microsomal monooxygenase similar to the one isolated from liver, the iodotransferring soluble factor, and an aminoethanethiol (RSH) tentatively identified as 4-phosphopantetheine. The system also requires oxygen [12], reduced pyridine nucleotides [13], and low concentrations of iodide. In contrast to the purported peroxide-dependent pathway, NADPH-dependent iodination is far less sensitive to glutathione and is quite specific for peptidyl substrates.
The ill-defined method of reconstitution used in this study probably underestimates the recovery of activity possible with these procedures, since incomplete recovery of limiting components diminishes the degree of synergism obtainable. Removal of an inhibitor, evidenced in both Table 1 and Table 5, also complicates estimates of recovery. Accurate assessment can only be made when all components of the system have been characterized and individually appraised throughout the purification procedures. Disproportionate increases of limiting components in the reconstitution can then balance deficits incurred during purification.
Several possible modulators of the NADPH-dependent pathway have also been identified. In accordance with other studies [13,26,27,28,29] calcium enhances metabolism of iodide and is a potential intracellular modulator of the NADPH-dependent iodinating pathway. Chelation of calcium by fully iodinated thyroglobulin may be important to maturation and coupling processes. Metabolism of 4-phosphopantetheine to cysteamine could accelerate maturation, since the monooxygenase and the smaller aminoethanethiol catalyze formation of proteinaceous disulfides [78,79,80]. Availability of 4-phosphopantetheine does seem to influence basal iodinating activity. Apparent deficiencies of the aminoethanethiol in this study are probably an artifact of purification procedures, but may reflect the regulatory and nutritional status of the intact animal.
HUMORAL MODIFIERS OF THYROID FUNCTION
Investigations of circulating levels of thyroxine, and of humoral factors influencing these levels, have established that the thyroid is essential to thermal homeostasis. In mammals the synthesis, storage, and release of hormones from the thyroid is controlled by the following cascade of events [4]:
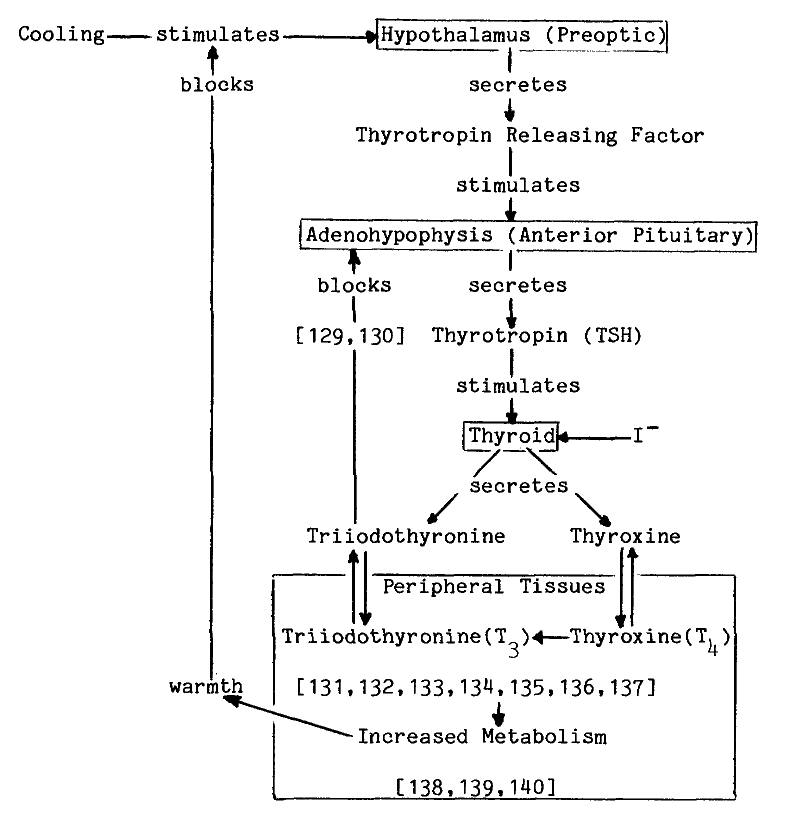
In perfect hindsight, extremely high fevers
would be expected in postoperative thyroid storm, when lesions in a hyperactive
thyroid dump stored thermogenic hormone into an already saturated system.
While useful in understanding physiological responses, this schematic grossly
oversimplifies the complex biochemical and biophysical interactions that
occur at each stage of the system. This review, concerned only with the
uptake and organification of iodide in the thyroid, merely introduces a
novel approach to understanding the thyroidal system.
UPTAKE OF IODIDE BY THE THYROID
The uptake of iodide by the thyroid is the subject of several reviews [29,31,39,75,76], and even greater controversy. Both the distribution of iodide in thyroidal pools and the biological significance of various purified and reconstituted systems are in question. These iodide-metabolizing systems often must be subjected to harsh treatments and nonphysiological conditions to obtain responses paralleling those seen in vivo and with intact tissue. Clearly intrafollicular concentrations of iodide are important to studies of the iodinating system.
Thyroidal Distribution of Iodine. Circulating concentrations of iodide fluctuate over a wide range, but seldom exceed 50 nM [2,19,141]. In contrast, thyroidal concentrations of iodine are normally 1 to 7 mM [29,75]. Of this accumulated iodine very little is iodide; most is found bound to thyroglobulin (80 to 90 percent), other proteins (0 to 15 percent), and to insoluble particulate material (10 to 15 percent). Iodide comprises 0.26 percent of the total iodine, or about 2 to 18 µM [3,75,142]. Refinements in technique indicate that up to 75 percent of this iodide results from deiodinations of previously organified iodide [40]. Furthermore, most of this iodide (99 percent) is not freely exchangeable with extracellular or recently accumulated iodide [3,19,75].
Thyrotropin (TSH) increases intrathyroidal iodide by enhancing the enzymic deiodination of tyrosyl iodine [40,141]. This may involve endocytosis of colloid [42], which is interesting considering the following electrochemical gradients established in the follicle [29]:
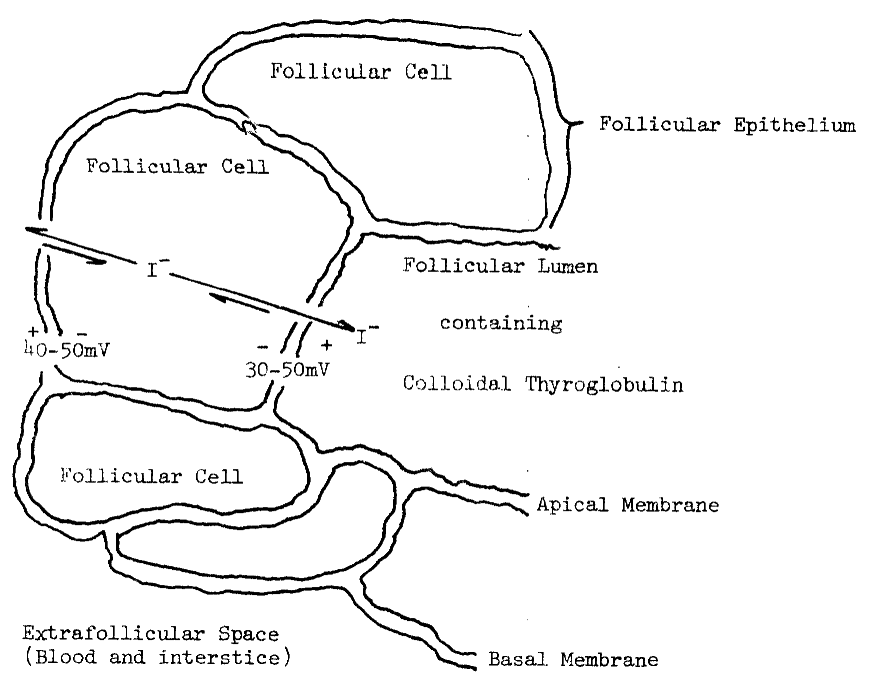
Iodide arising in follicularized cells may
sequester into the colloid along the apical gradient [76,143],
and return to the blood across the basal membrane. Thyroglobulin (Tg) strengthens
the gradient into the lumen by displaying a low affinity for iodide [31].
Poorly exchangeable iodide [3,19,75]
may be retained by gradients into the lumen, while freely exchangeable
iodide resides in the follicular cell (20 to 180 nM). Much of the iodide
is, in fact, located within the colloid [19,39].
Freely exchangeable iodide is discharged by perchlorate, but the latter
anion has not been localized. Consequently, this distribution of iodide
cannot be confirmed. Alternately, the poorly exchangeable pool may arise
from deiodination of an organic compound not discharged from thyroidal
tissue by perchlorate [75]. Regardless of its source,
concentrations of freely exchangeable iodide within the cell may dip below
that of serum.
Requirements for Uptake. Oxygen is essential for the accumulation of iodide by the thyroid [11,19,31,39]. As much as 40 percent of the total respiration in thyroid slices is directed towards the production of ATP for a Na+-K+ requiring ATPase [29,31]. The biphasic response of respiration to increasing oxygen tension may indicate multiple pathways for oxygen. One component accounts for roughly 40 percent of the total respiration, and saturates at 2 to 5 percent oxygen. A second component is only half-saturated over this range. Respiration and accumulation of iodide vary concordantly at higher oxygen tensions, 5 to 100 percent [11].
Accumulation of iodide and respiration vary similarly with changes in pH, but not with temperature. Respiration and accumulation are optimal at pH 7.5 to 8.0 in slices [11], and at pH 7.0 in homogenates [61]. These values compare with the postulated pHs, 7.4 and 7.0, of their respective compartments, blood and cytosol [30,31]. Accumulation of iodide is optimal in slices at 38°C [11], a temperature that inactivates homogenates [61], but drops to half-maximal at 45°C, the optimum for respiration. Cooling and anoxia decrease respiration and the accumulation of iodide in slices, while warming and oxygen restore both activities to original levels [11].
Sodium, potassium, and calcium ions, but not
magnesium ions, are required for maximal accumulation of iodide. Concentrations
of calcium, 0.9 mM [29], approaching that of serum, 1.16
mM [4,30], are optimal. No explanation
is currently available for the calcium requirement. It is noteworthy, however,
that calcium iodide is quite soluble in alcohol and acetone, producing
a cationic form of iodide. Sodium is probably most effective in the media,
since its effects on the accumulation of iodide are immediate. Potassium
must first equilibrate with media or tissue, and is probably most effective
within some compartment of the follicle [29]. This agrees
with the usual mammalian distribution of sodium and potassium, and with
the unidirectional activity of the Na+-K+
ATPase; sodium is pumped out as potassium is pumped into the cell [4,144-147].
Evidence for Passive Diffusion. Concentrations of intracellular iodide roughly equal to those normally found in blood (50 nM) indicate passive diffusion of iodide across the basal membrane. The iodide diffusion space accounts for a relatively large portion of the total thyroidal volume (35 percent). As might be expected in a pore-mediated equilibration, the smaller anion, chloride, has approximately the same diffusion space [39]. The larger anion, perchlorate, does not influence iodides space [31,75]. Experimental modification of the follicular lumen and its contents without effect on the diffusion space [39] indicates that the lumen is not involved.
The diffused iodide is available for organification [39]. The rate of organification of iodide in the thyroid is estimated to be from 90 percent per hour [148] to 190 percent per minute [75], or instantaneously [19,39,148]. Unfortunately, cell-free demonstration of organification is extremely difficult, leading many to discount its significance in the accumulation of iodide [29,39]. Homogenates of thyroid, nonetheless, accumulate iodide. This activity, unlike accumulation by intact cells, is quite labile at physiological temperatures. Additional incongruent effects are observed in response to propylthiouracil treatment; accumulation of iodide is decreased in homogenates but increased in cells [61,75]. The most plausible explanation for these discrepancies is that dialysis tubing is much more permeable to stabilizing factor(s) and intermediates than an intact membrane. Even though a metabolic gradient for iodide into the thyroid exists, its contribution to the accumulation of iodide cannot be ascertained with currently used probes. Indeed, use of the uncoupling agent, propylthiouracil, leads to conflicting views of the concentrating mechanism.
Clinical evidence is also used to belittle the import of passive diffusion and subsequent organification in the concentration of iodide. Goitres arising from a defect in the concentrating mechanism have been identified. Since only high doses of iodide alleviate the problem, diffusion is presumed to be of consequence only at superphysiological concentrations of iodide [29,31]. Clearly, until the genetic lesion is identified in these individuals, and until the concentrating process is better understood, such conclusions are unfounded.
Evidence for Active Transport. Low concentrations of freely exchangeable, intrathyroidal iodide are not observed in tissues or cells treated with certain thiocarbamides. Tissues pretreated with propylthiouracil [19,29,31,39,75] and isolated cells incubated in methimazole (2-mercapto-N-methylimidazole) [141] accumulate much more freely exchangeable isotope. Fifty to sixty-fold concentration is possible, when medium contains 1 µM iodide. Tissues and isolated cells are similarly saturated by 0.1 mM iodide [29,141]. Total accumulation of iodine is considerably less than the 1 to 7 mM observed in untreated tissue [29,75]. These two antithyroidal agents are presumed to block organification without affecting transport of the anion. Hence, iodide accumulates in the perchlorate-labile pool as the following scheme illustrates [19,29,31,39,75,141]:
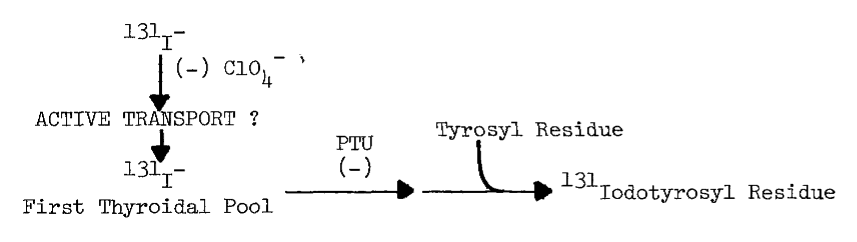
This interpretation is incongruent with other observations. Isotopic iodide accumulates more rapidly [75], but equilibrates with total thyroidal iodine more slowly in propylthiouracil-treated tissues [29] than in untreated tissues [39]. Apparently, mixing of iodine isotopes is prevented by propylthiouracil. Although this mixing involves organification and deiodination [75], the effect is generally assumed to be on the former. The following scheme illustrates the complex interactions observed in the presence of propylthiouracil:
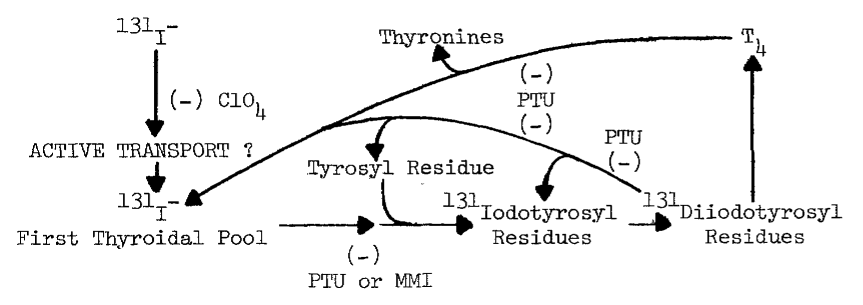
Propylthiouracil is known to block the deiodination of thyroxine in the thyroid [149] and peripheral tissues [132,133,150]. It also appears to depress the deiodination of previously organified sources by up to 87 percent [42]. Increases in specific activity are easily misinterpreted as increases in concentrations, since tracer iodide is diluted less in propylthiouracil-treated tissues. Lower specific activities of radioiodine have been observed in untreated tissues [38]. The import of active transport relative to passive diffusion is, consequenty, exaggerated when preparations are pretreated with propylthiouracil.
Methimazole (MMI), in contrast to thiouracil, effectively depresses formation of iodotyrosine and iodothyronines [11] with relatively little inhibition of deiodination [151,152]. An increase in exchangeable iodide with methimazole treatment [29,141] argues for the existence of actively transported iodide; methimazole treatment produces greater accumulation of radioiodine [76] regardless of the propensity towards higher specific activities of iodine isotopes when propylthiouracil is used. The accumulated iodine is indistinguishable from iodide with currently used techniques [31,76].
Propylthiouracil and methimazole do not affect respiration in the thyroid, even at concentrations greatly exceeding those necessary to block organification [11]. One must assume that (1) the system for organification is saturated with iodide, (2) that these thiols do not directly compete with iodide for oxidative equivalents, or that (3) organification requires an insignificant portion of the total respiration. The first assumption is false, since accumulation of iodide is optimal at superphysiological concentrations of the anion [19,39,75,148]. The second point is possible, but products in the presence of thiocarbamides have not been identified. The last, (3), remains an alternative even though the systems reponsible for most of the respiration have not been identified [11,29,31]. The respiration lost from the tissue soon after removal from the animal may be most important to the accumulation of iodide. Respiration drops at least 15 percent during processing at the abattoir, transportation, and technical preparation in the laboratory. Accumulation of iodide declines even more precipitously with aging than does respiration [11].
The thyroid concentrates certain other anions with a concommitant loss of iodide. These anionsClO4, TeO4, and ReO4are thought to compete with iodide for a carrier in the basal membrane. It follows that they accumulate instead of iodide [29,31]. Alternately, accumulation of these anions may interfere with a facilitated diffusion of iodide along the apical gradient. Radioautographic localization of iodide in propylthiouracil-treated tissues supports the former or active transport theory; iodide accumulates throughout follicular cells and later migrates into the lumen [143]. In untreated tissues iodine is limited to the apical membrane and vicinal colloid [19,153], and does not permeate the cell.
Concentration of Analogous Anions. Those anions most effective in inhibiting the uptake of iodide typically (a) are monovalent, (b) have larger partial molal ionic volumes that iodide, (c) are tetrahedral rather than planar or linear, and (d) are not metabolized by the thyroid. These are generally regarded as size and shape requirements for a hypothetical carrier. A choline and nervonic acid containing phospholipid may be the carrier [29,31,76]. A cephalin displaying even greater specificity and capacity for iodide has not been fully characterized, as it is relatively labile [154]. Alternatively, these observations may define an ionic pore that screens anions for proper size, shape, and charge characteristics [31].
Thiocyanate is a particularly interesting inhibitor of iodide accumulation. Of the monovalent anions tested, this agent most closely resembles iodide in size [31]. SCN is linear, however, and consequently has a somewhat smaller transverse area. In contrast to a larger analogue, SeCN¯, SCN¯ is only slightly concentrated by the thyroid. A smaller analogue, OCN¯, is only weakly inhibitory [31]. Most of the SCN¯ accumulated, like iodide, is found in the lumen of follicules [155].
Failure to observe large accumulations of thiocyanate can be explained by the rapid metabolism of this agent in the thyroid [31]. The same enzymes catalyzing the metabolism of iodide may be involved [39], since either anion inhibits the metabolism of the other [31,61]. Metabolism of thiocyanate, like iodide, is increased by TSH and decreased by a member of the thiocarbamide class. In addition, both TSH and the thiocarbamide enhance thyroidal accumulation of radioactivity derived from thiocyanate, Though of lesser magnitude, these effects are similar in direction to those observed with iodide [156]. Thiocyanate is oxidized in the following reaction catalyzed by a variety of peroxidases [157]:

The sulfenate produced, can only be generated enzymically, and decomposes by the following postulated mechanism to form sulfate and cyanate [157]:
![]()
In contrast to metabolites of iodide, sulfate is not concentrated by the thyroid [31,156], so lesser accumulation of label from thiocyanate is expected.
Other observations attest to similarities in
transport for these two anions. Accumulated iodide is rapidly discharged
from the thyroid by thiocyanate, even though uptake of the latter is slight,
and even when the accumulation of iodide is favored by incubation with
propylthiouracil [31]. Furthermore, increasing iodide
to 30 to 50 µM, or adding 20 to 60 µM thiocyanate half-suppresses
the uptake of iodide from media containing 1 µM iodide [29,31,141].
The deleterious effect of iodide upon its own accumulation is described
in greater detail later, as are other analogous actions of thiocyanate.
FACTORS INFLUENCING IODIDE'S ACCUMULATION
Table 9 is a summary of endogenous factors influencing the accumulation of iodide, and of elapsed times after each stimulus until the effect is observed.
TABLE 9
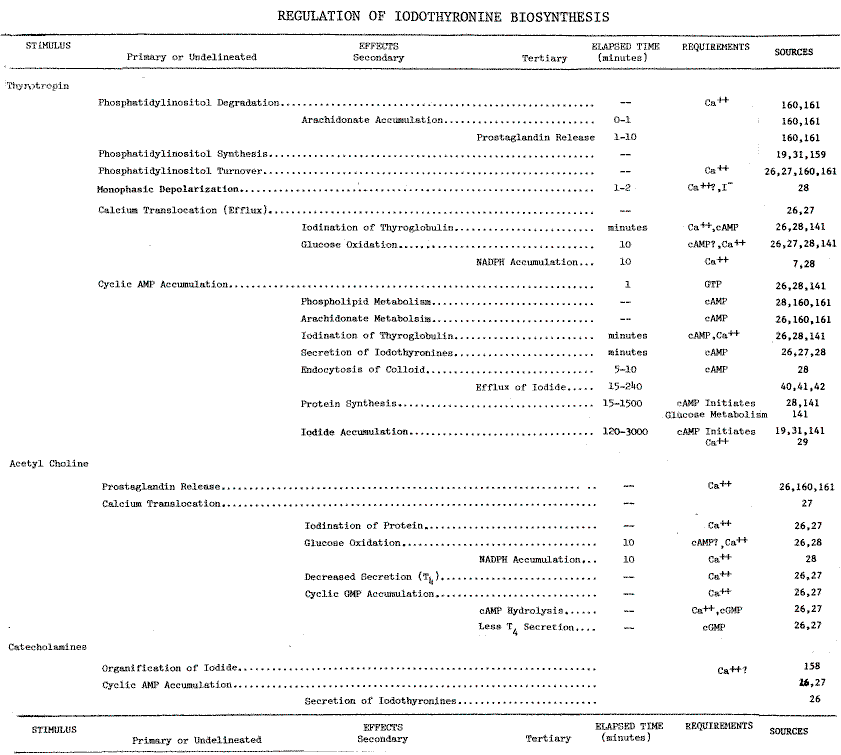
Catecholamines. Norepinephrine and epinephrine stimulate thyroidal uptake and organification of iodide [158], even in the presence of the purported transport inhibitors [31]: ouabain, digitoxin, and perchlorate. Iodine apparently accumulates along gradients established by organification, since blocking organification with methimazole prevents stimulation [158].
Acetylcholine. Responses to acetyleholine appear to be more species-specific. In thyroids of dogs, acetyleholine elicits a rapid rise in intracellular calcium. Effects secondary to a rise in calcium include (1) accumulation of cGMP, (2) oxidation of glucose, (3) iodination of protein, and (4) release of prostaglandins. Acetyleholine suppresses TSH-induced accumulation of cyclic AMP in dog thyroids. In contrast, cyclic AMP accumulates in the human thyroid regardless of exposure to acetylcholine. and subsequent rises in intracellular calcium and cyclic GMP [26,27].
Thyrotropin. TSH is the most important endocrine modulator of the thyroid. Responses to this hormone can be conveniently categorized as early or late (Table 9). Early responses include (1) accumulation of cyclic AMP, (2) oxidation of glucose, and (3) iodination of protein [141]. Of particular interest is the release of iodide from preloaded thyroids. This response to TSH is augmented by perehlorate and propylthiouracil, that prevent uptake and organification of liberated iodide [41]. The efflux is thought to stem from deiodination of previously iodinated thyroglobulin [31], and coincides with cAMP-dependent endocytosis of colloid and subsequent proteolytic liberation of iodothyronines [19,26,27,28]. Proteolysis and NADPH-dependent deiodination are optimal after 2 and 3 hours, respectively [28].
Thyrotropin also elicits a rapid increase in the activity of the Na+-K+ ATPase. The system is thought to be involved in the accumulation of iodide [31], accumulation is enhanced only at much later times [19,31,141]. Optimal accumulation is not observed for days. Another delayed response (Table 9), efflux, allegedly masks accumulation at early times [31]. Protein synthesis is also increased by TSH at a later time. These proteins are supposedly are linked to the active transport of iodide [19,141], but treatment with triiodothyronine or hypophysectomy depresses accumulation of iodide without influencing Na+-K+ ATPase activity [28]. While the ATPase may be necessary for the accumulation of iodide, it is not apparently sufficient.
Thyrotropin is known to specifically enhance the synthesis of thyroglobulin (19S) and its 12S precursor. As with the accumulation of iodide, increases in thyroglobulin are obvious at later times, after 24 hours [28]. An attendent increase in the gradient for iodide into the lumen [31] could account for much of the TSH-dependent accumulation of iodide.
Another early response is a stimulation of phospholipid synthesis, particularly phosphatidylinositol [19,31, 159]. Thyroidal phosphatidylinositol is particularly enriched in arachidonic acid, precursor of prostaglandins [160,161].
Other Endocrine Modulators. Iodothyronines, in an apparent feedback
loop, also influence the uptake of iodide. Triiodothyronine lowers, in
vivo, the intrathyroidal concentration of iodide [39],
but neither triiodothyronine nor thyroxine influences the accumulation
of iodide in isolated cells [45]. Similarly, thyroid
hormones have no effect on TSH-induced accumulation of cyclic AMP by isolated
cells [46]. Triiodothyronine and, to a lesser extent,
thyroxine do suppress the secretion of TSH by the adenohypophysis [129,130].
Thyroid hormones have little immediate effect upon the thyroid [26,45].
Xenobiotic Compounds. Iodide-dependent destruction of reduced pyridine nucleotides is also observed in vivo in rat thyroids [53]. Unlike cysteamine, propylthiouracil and methimazole prevent formation of iodinated tyrosyl residues [127], without protecting NADH and NADPH [47,53]. In contrast to thiocarbamides, perchlorate completely protects endogenous pyridine nucleotides from destruction at high concentrations of iodide [53].
Propyithiouracil and methimazole also elicit thyroidal accumulation of iodide [19,29,31,39,75,76]. When organification is minimized by high concentrations of iodide, though, propylthiouracil depresses accumulation of the isotope [39]. Iodide, acting alone, is thought to divert oxidized intermediates from the hormonogenic pathway [48]. Results in Table 8 indicate that 1) the intermediate may be a sulfenyl iodide formed from an aminoethanethiol, and 2) that thyroid peroxidase catalyzes the iodide-dependent destabilization of this intermediate. Propylthiouracil may potentiate the uncoupling by removing the aminoethanethiol or by producing a nonhormonogenic sulfenyl iodide. Since propylthiouracil and methimazole are concentrated by the thyroid 500 and 1000 fold, respectively [33,34], considerable accumulations of their sulfenyl iodides are possible. These sulfenyl iodides probably have the usual bond polarization, and should be more susceptible to nucleophilic substitution of iodine than sulfenyl iodides derived from aminoethanethiols. Thiocarbamides in the thyroid may, therefore, constitute the reported system for accumulating iodide [29,31,39,75,76].
Action of thiocyanate in the system is probably analogous to iodide. Although highly reactive, sulfenyl thiocyanates are formed, and behave in a manner similar to sulfenyl iodides [77]. Competition with iodide for the sulfenic acid could account for the antithyroidal potency of thiocyanate. Like the iodines, sulfenyl thiocyanates may form at low concentrations of anion (Figure 10) without appreciably influencing the uptake of oxygen. Formation of reactive sulfenyl thiocyanates may be important to a related thyroidal activity. The amount of thiocyanate half-inhibiting NADPH-dependent iodination, 83 µM (Figure 11), is similar to the concentration half-stimulating the coupling reaction catalyzed by thyroid peroxidase, 0.1 mM. As noted earlier, the effectiveness of thiocyanate in stimulating the coupling reaction depends upon the tyrosyl substrate used; much lower concentrations are required with the native substrate, thyroglobulin [162].
Peroxide-dependent iodination catalyzed by thyroidal microsomes is also inhibited by thiocyanate, but only at higher concentrations; at lower concentrations, thiocyanate enhances this iodination of half-cystinly P-9. Stimulation apparently is dependent upon the availability of peroxide, since increasing the rate of peroxide generation also increases the rate of iodination (Figure 10). Although thiocyanate can be used to differentiate between peroxide-dependent and NADPH-dependent iodination, glutathione is an effective endogenous determinant (Figure 9, Table 4).
Sensitivity of NADPH-dependent iodination (Figure 10) and of the accumulating system to thiocyanate suggests a common pathway. Although speculative at this time, inhibition of monooxygenase-catalyzed reactions by thiocyanate indicates involvement of this enzyme in the metabolism of iodide.
VITA
Galen Daryl Knight was born in Emporia, Kansas, on February 23, 1954, the son of Daryl C. and Ladonna J. Knight. After completing his work at Lebo High School, Lebo, Kansas, in 1972, he entered Southwestern College in Winfield, Kansas, where he received the Bachelor of Science degree with majors in Chemistry and Bio1ogy in May of 1976. Shortly thereafter he entered the Graduate School of the University of Texas at Austin.Permanent address:
Rural Route 2 Lebo, Kansas 66856This dissertation was typed, in part, by Beth Knight and Nancy Ligrani.
| Home | Overview | People | Journal | Nutrition |
| Environment |
|
WWW Links | Outline | e-mail us |